

I februari går patenten ut för de två storsäljande TNF-alfahämmarna Remicade (infliximab) och Enbrel (etanercept) i Sverige och stora delar av EU. Och lite senare i vår går patentet ut för diabetesmedicinen Lantus (insulin glargin).
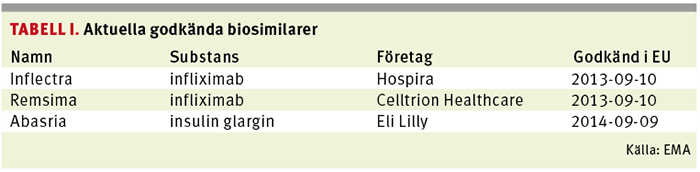
Då kan de biologiska kopiorna – biosimilarerna – på infliximab (Inflectra och Remsima) och insulin glargin (Abasria) som redan finns godkända hos den europeiska läkemedelsmyndigheten EMA börja säljas.
Precis som med generiska läkemedelskopior är det främsta argumentet för biosimilarer priset, men det handlar inte alls om samma dramatiska prissänkningar som är vanligt vid generisk konkurrens, där priserna kan falla med 95 procent. Erfarenhetsmässigt har priserna på biosimilarer i stället landat någonstans mellan 15 och 35 procent under priset på originalet.
Just nu ligger företagen och lurpassar på varandra, och de officiella priserna hos Tandvårds- och läkemedelsförmånsverket, TLV, är nästan identiska där priserna för biosimilarerna ligger mindre än två procent under originalet Remicade.
Av patenttekniska skäl vill företagen som marknadsför biosimilarerna inte uttala sig alls kring sina strategier eller målsättningar, men det går att titta på utvecklingen i Norge där de biologiska kopiorna på infliximab har funnits i cirka ett år redan. Där ligger priset på den billigaste biosimilaren 39 procent under originalet Remicade, och den har hittills tagit 33 procent av infliximab-marknaden i Norge.
Med tanke på att Remicade är Sveriges största slutenvårdspreparat och kostade svenska landsting strax under 600 miljoner kronor 2014, enligt E-hälsomyndigheten, så kan besparingen för landstingen bli betydande om samma utveckling sker här. Detsamma gäller för Lantus som förskrevs för knappt 240 miljoner i öppenvården förra året.
För den andra TNF-alfahämmaren som förlorar sitt patent nu i februari, Enbrel, som såldes för knappt 800 miljoner kronor 2014, finns det ännu ingen godkänd biosimilar. EMA meddelade dock i början av januari att man har fått in en ansökan om ett godkännande, så det är en på gång.
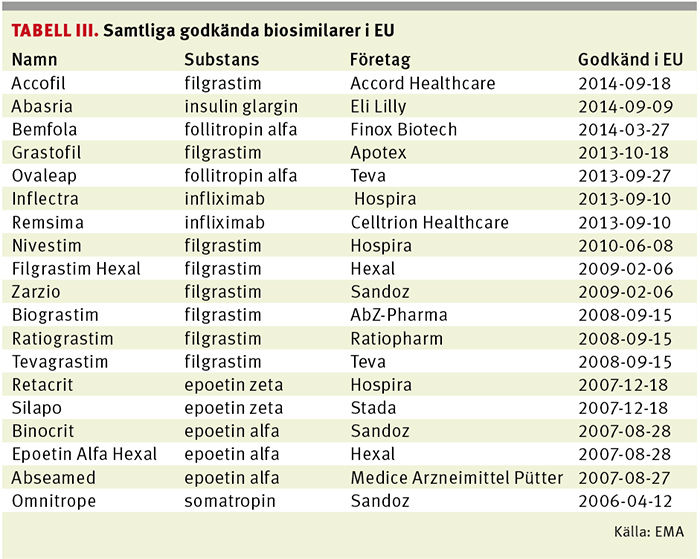
Biosimilarer är egentligen inget nytt. De har funnits godkända inom EU, och därmed även i Sverige, sedan tillväxthormonet Omnitrope, innehållande somatropin, godkändes 2006. I dag finns det knappt 20 godkända biosimilarer inom EU innehållande bland annat epoetin, filgrastim och folitropin.
Biosimilarer är inte detsamma som generiska kopior av biologiska läkemedel. De är visserligen ett slags kopior, men eftersom det handlar om biologiska läkemedel är begreppet kopia lite mer komplicerat än för vanliga läkemedel.
Biologiska läkemedel, som de monoklonala antikropparna till exempel, är betydligt större och mer komplexa molekyler än vanliga läkemedel. Jämför den kemiska summaformeln för paracetamol – C8H9NO2 – eller för morfin – C17H19NO3 – med summaformeln för infliximab – C6428H9912N1694O1987S46 – så blir det uppenbart. Därför blir också kontrollen för att avgöra om kopian är tillräckligt lik originalet betydligt mer komplicerad än för vanliga generika. Det är nämligen inte bara antalet atomer som måste vara korrekt, molekylen måste dessutom ha den rätta tredimensionella strukturen och den måste modifieras i kroppen på ett liknande sätt som originalet.
Biosimilarer jämförs därför med originalpreparatet dels i en omfattande fysikalisk och biologisk karaktärisering av exempelvis molekylstruktur, receptorbindning och biologisk aktivitet, dels i studier av farmakokinetik, farmakodynamik och immunogenitet.
– Det är väldigt omfattande undersökningar, och det tror jag kanske att kliniker ibland underskattar. Därför blir de ibland lite undrande kring varför de kliniska studierna till sitt omfång är mer begränsade jämfört med originalpreparatet, säger Bengt Ljungberg, senior vetenskaplig rådgivare vid Läkemedelsverket.
För biosimilarer behöver trots allt inte genomgå kliniska studier i samma utsträckning som originalpreparatet har behövt. Det räcker oftast med att biosimilaren har visats ha effekt vid en eller två indikationer för att den ska bli godkänd på samtliga av originalpreparatets indikationer.
– Ja, det är grundprincipen, annars vore ju konceptet lite meningslöst. Det bygger på att man vilar på den fysikaliska, biokemiska karaktäriseringen och att man driver den så pass långt som man gör. Grundprincipen är att man får samtliga indikationer som originalpreparatet, säger Bengt Ljungberg.
De båda infliximab-preparaten Inflectra och Remsima är så kallade duplikat. Det innebär att det egentligen är samma produkt, godkänd på exakt samma dokumentation, men med två olika företag som marknadsför den under olika namn. Den kliniska dokumentationen är endast gjord på reumatoid artrit och ankyloserande spondylit, men de båda biosimilarerna har fått Remicades samtliga åtta indikationer.
Denna extrapolering av indikationer är kontroversiell inom läkemedelsmyndighetsvärlden; europeiska EMA går längst, medan den kanadensiska myndigheten Health Canada i stället starkt avråder från att extrapolera indikationer och också bara har godkänt infliximab-biosimilarerna på de två indikationer där det finns klinisk dokumentation. Amerikanska FDA ligger i detta sammanhang långt efter de övriga myndigheterna, och tveksamheten kring biosimilarer har varit stor. Strax efter årsskiftet rekommenderade dock myndighetens rådgivande läkemedelsnämnd att den första biosimilaren i USA, filgrastim, skulle godkännas på originalets samtliga fem indikationer trots att den endast hade klinisk dokumentation på en av dem, men något beslut är ännu inte fattat. Hur den amerikanska myndigheten ställer sig i frågan om indikationsextrapolering är därför ännu inte klart. Men att det är en sådan skillnad mellan olika godkännandemyndigheters syn på biosimilarer är inte särskilt konstigt, menar Bengt Ljungberg.
– Det förekommer ju i många sammanhang att vi, FDA och Kanada kommer till lite olika bedömningar. Så detta förvånar mig inte, säger han.
Bengt Ljungberg menar dock att myndigheten i Kanada är väl försiktig och inte litar på sin egen bedömning av preparatens likhet.
– Det kan, som en generell princip, ses som en logisk kullerbytta, i mitt tycke, säger han.
Men samtidigt framhåller han att detta är en fråga som måste hanteras från läkemedel till läkemedel. Den monoklonala antikroppen rituximab (Mabthera) har lyfts fram som extra komplicerad när det gäller indikationsextrapolering eftersom den både har indikationen reumatoid artrit och non-Hodgkin-lymfom, med helt olika patientgrupper.
– Det är ett bra exempel där frågan kan ställas på sin spets. Då får vi ju se var vi hamnar någonstans. Indikationsextrapolering fordrar en individuell bedömning, man kan inte fastställa att det alltid ska vara på ett visst sätt, men som EMA hittills har sett det så har det varit grundprincipen, säger Bengt Ljungberg.
Eftersom kraven på klinisk dokumentation är lägre för biosimilarer än för originalprodukten är det naturligtvis viktigt att uppföljningen är god.
– Utgångspunkten är att det ska vara samma krav på biosimilaren som på originalet, sedan kan det ju tillkomma någonting om det finns skäl för det, säger Bengt Ljungberg.
I de aktuella fallen finns inga ytterligare krav på uppföljning. I själva verket är kraven på de båda infliximab-preparaten Inflectra och Remsima faktiskt mindre omfattande än de har varit för Remicade. Men det handlar mer om att TNF-alfahämmare var något helt nytt 1999 när det godkändes, och därför har myndigheterna ställt höga krav på uppföljningen, även när nya indikationer har godkänts.
– För Remicade har det funnits krav på ett ganska omfattande program av ytterligare studier. När den godkändes var detta ett väldigt okänt område, säger Ulla Wändel Liminga, ämnesområdesansvarig för farmakologi och toxikologi på Läkemedelsverket och svensk ledamot i EMA:s Pharmacovigilance Risk Assessment Committee, PRAC.
Riskhanteringsplanen för Inflectra och Remsima följer i huvudsak standardmallen med signalspaning i databaser, periodiska säkerhetsrapporter och litteraturföljning. Dessutom finns krav på uppföljningsstudier inom reumatologi och gastroenterologi samt uppföljning i de reumaregister som redan finns i olika europeiska länder, som det svenska.
Inflectra och Remsima har redan funnits i cirka ett år i europeiska länder som Norge, Finland, Portugal och Tjeckien, och ingenstans har det kommit några signaler om att de uppträder annorlunda än originalet.
– EMA skannar varannan vecka igenom biverkningsdatabasen efter biverkningar. Det är visserligen ett ganska trubbigt verktyg, och en avsaknad av signal i biverkningsdatabasen betyder ju inte att det inte kan finnas problem, men det är ju bättre att vi inte har hittat något än om vi hade gjort det, säger Ulla Wändel Liminga.
Ett problem när det gäller uppföljning av biologiska läkemedel, oavsett om det handlar om biosimilarer eller original, är de bristfälliga möjligheterna att följa preparat från olika tillverkningsbatcher. För precis som biosimilarer aldrig kan vara identiska kopior av originalen så kan inte heller preparat från två batcher av samma biologiska läkemedel vara identiska.
Därför har det sedan den nya farmakovigilanslagen infördes i juli 2012 funnits ett myndighetskrav från EMA på att inte bara läkemedelsnamnet utan även batchnumret ska registreras i patienternas journaler för alla biologiska läkemedel. Problemet är bara att det inte görs i någon högre utsträckning.
Enligt Ulla Wändel Liminga har Läkemedelsverket lyft frågan till EU-nivå om det inte borde utvecklas ett verktyg för att göra det möjligt att följa kraven, men det har ännu inte resulterat i något.
– Det är bara att erkänna att det finns ett glapp här, säger hon.
Samtidigt menar Bengt Ljungberg att situationen kan hanteras.
– Vi har stor erfarenhet från andra sammanhang när man ska spåra batcher och det har ju i praktiken sällan varit något stort problem. Så jag är inte så orolig för vad som skulle kunna tänkas hända, säger han.
Anledningen till att inget hänt kan vara att läkemedelsföretagen håller på att utveckla ett system för att skydda Europas läkemedelsdistribution från förfalskade läkemedel. Där kommer varje expedierad läkemedelsförpackning att vara spårbar med batchnummer och allt.
– Det kommer förhoppningsvis att lösa även denna fråga, säger Bengt Ljungberg.
I motsats till generika, som i regel är utbytbara på apotek, så är biosimilarer i Sverige inte utbytbara. En av orsakerna är att man inte vill utsätta patienterna för upprepade byten av biologiska preparat med små skillnader som teoretiskt skulle kunna öka risken för immunreaktioner med autoantikroppsbildning som följd. Frågan ligger på de nationella myndigheterna att besluta om, och Bengt Ljungberg ser inte att det kommer att ändras.
– Vi har hittills inte ansett det rimligt att de ska vara utbytbara. Det är inte särskilt kontroversiellt i ett svenskt perspektiv, säger han.
I Norge är biosimilarer inte heller utbytbara på apotek. Däremot börjar allt fler sjukhus nu vilja ställa över sina patienter från originalpreparatet Remicade till en infliximab-biosimilar av ekonomiska skäl, något som har orsakat en viss diskussion och resulterat i att den norska staten har finansierat en studie där de ska jämföra 300 reumapatienter som ställs över på Remsima med lika många som står kvar på Remicade.
Men i Sverige skulle en liknande utveckling inte vara något bekymmer för Läkemedelsverket, menar Bengt Ljungberg.
– Om man generellt på en klinik bestämmer sig för att gå över till en biosimilar så kan ju inte jag utifrån mitt perspektiv se något problem med det. Ur ett regulatoriskt perspektiv skulle vi inte ha något att invända mot det, men det är nog bra om man har professionen med sig i så fall, säger han.