






Torakala aortaaneurysm och dissektioner kan förekomma som en isolerad manifestation, som en del av ett känt syndrom eller som en ärftlig sjukdom utan koppling till kända syndrom.
I ca 20 procent av de familjära fallen kan en genetisk avvikelse identifieras.
Sjukdomen orsakas av en dysfunktion hos de glatta muskelcellerna eller i stödjeproteinerna i torakala aortaväggen.
Vid de ärftliga formerna kan dissektion i torakala aorta inträffa vid mindre diameter än i de sporadiska fallen.
Angiotensinreceptorblockerare (ARB) förefaller ge en långsammare aneurysmtillväxt än placebo. Jämförande studier mellan ARB och betablockerare pågår.
Patogenesen vid torakala aortaaneurysm (TAA) skiljer sig i allmänhet från den vid abdominella aortaaneurysm (AAA). Medan AAA är nära kopplade till aterosklerotiska riskfaktorer är bakgrunden till TAA oftare en defekt funktion hos glatta muskelceller eller bindvävsproteiner [1]. Aneurysm i aorta descendens och de torakoabdominella aneurysmen kan ha båda dessa bakgrunder.
Den årliga incidensen av torakala aortaaneurysm och dissektioner (TAAD) – sjukhusvårdade och döda – i Sverige är ca 9/100 000 kvinnor och 16/100 000 män [2]. Ungefär hälften av dessa utgörs av aortadissektioner. Ett mörkertal finns då ett antal individer avlider utan att diagnos fastställs. Under år 2011 utfördes sammanlagt 961 (elektiva och akuta) operationer på torakala aorta i Sverige. Antalet operationer har tredubblats under ett drygt decennium (Figur 1).
Aortadissektion är ett livshotande tillstånd som kräver omedelbart omhändertagande. Vid dissektion sker en bristning av insidan av aortas vägg varvid blod strömmar in i mellersta lagret, media, och klyver aortaväggen i två lager. Flöde uppstår då i två lumen; förutom i det »äkta« lumen också i ett »falskt« lumen (Figur 2). Återflödet tillbaka till aortas äkta lumen kan ske på olika ställen under aortas förlopp mot bifurkationen till benartärerna.
Det vanligaste symtomet vid aortadissektion är kraftig bröst- eller ryggsmärta som ibland kan vara migrerande. Övriga symtom härrör i regel från de organsystem som drabbas till följd av dissektionen; hjärtinfarkt om dissektionen når kranskärlen, cirkulatorisk chock om dissektionen utvecklas till en hjärttamponad, stroke eller andra neurologiska symtom om dissektionen påverkar kärlavgångarna till centrala nervsystemet. Dissektion som ockluderar kärlavgångar till bukorganen ger buk- eller ryggsmärtor, och längre distalt ses ibland även akut extremitetsischemi. Denna mångfacetterade bild leder inte sällan till fördröjd diagnos eller till att den kliniska bilden misstolkas som en trombotisk ocklusion som behandlas med t ex trombolys.
Dissektioner som drabbar den uppåtstigande delen av aorta (aorta ascendens), typ A-dissektioner, behandlas med omedelbar operation om patientens tillstånd så tillåter. Dissektioner som drabbar den nedåtstigande delen av aorta (aorta descendens), typ B-dissektioner, behandlas i regel konservativt med rigorös kontroll av blodtrycket (Figur 3).
Familjära TAAD utan syndrom
Ca 20 procent av alla som drabbas av en aortadissektion har en släkting med dissektion eller aneurysm i torakala aorta [3]. I en del familjer är aortasjukdomen ett delfenomen i ett syndrom men oftast finns inget känt engagemang av andra organsystem.
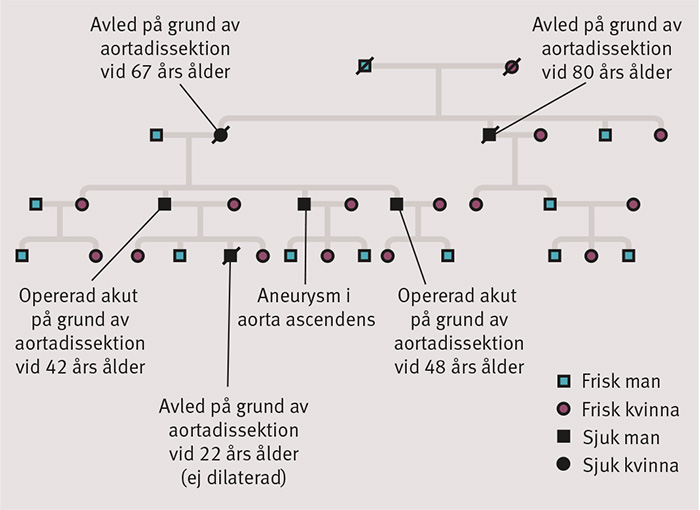
Nedärvningsmönstret är autosomalt dominant men visar både nedsatt penetrans (alla anlagsbärare utvecklar inte sjukdomen) och variabel expressivitet (varierande grad av symtom). Män drabbas oftare och tidigare av dissektioner än kvinnor. Dissektionen uppträder i regel i yngre åldrar och vid mindre diameter i de familjära fallen jämfört med i de sporadiska fallen [3]. Ett exempel på hur sjukdomen kan manifestera sig i en familj visas i Figur 4. Hos yngre individer kan dissektionen inträffa vid normal eller nära normal diameter. Avsaknad av andra organmanifestationer gör det då svårt att ställa diagnos innan dissektion inträffar. Det är också närmast omöjligt att utesluta sjukdomen hos yngre individer så länge den sjukdomsorsakande mutationen i familjerna inte är känd.
Familjära TAAD som del i syndrom
Flera ärftliga bindvävssyndrom är associerade med nedsatt hållfasthet i aorta och större artärer. Ärftlighetsgången vid dessa tillstånd är vanligtvis autosomalt dominant.
Det är lätt att förbise att det kan handla om ett ärftligt tillstånd, då anlagets penetrans inte är fullständigt och expressiviteten är varierande. Sjukdomen behöver inte heller vara nedärvd, då mutationen kan uppstå de novo, det vill säga som en nymutation hos individen.
Det finns i dag drygt 15 olika kända genetiska tillstånd som kan delmanifesteras i aortasjukdom. De vanligaste är Marfans syndrom, Ehlers–Danlos syndrom och Loeys–Dietz syndrom.
Marfans syndrom. Marfans syndrom är den vanligaste av de kända genetiska orsakerna till dissektion i proximala aorta. Syndromet orsakas av en mutation i FBN1–genen som kodar för fibrillin, ett extracellulärt matrixprotein som ingår i mikrofibriller och bidrar till vävnaders hållfasthet och till aortas eftergivlighet. Prevalensen anges till 1/5 000–10 000, och när syndromet diagnostiseras är en nymutation orsaken hos en tredjedel av individerna. Syndromet karakteriseras av långsmal kroppsbyggnad med långa smala armar, ben, fingrar och fötter. Dilatation av aortaroten är vanligt förekommande och dissektionen ses i regel före 40 års ålder. Även läckage i aortaklaffen och framför allt mitralisklaffen är vanligt. Övriga symtom är synnedsättning beroende på linsluxation, spontanpneumotorax och ryggradskrökning. Gravida kvinnor med Marfans syndrom löper ökad risk för aortadissektion. Profylaktisk aortakirurgi före graviditet bör övervägas redan vid lätt dilaterad aortarot.
Marfans syndrom är en kriteriediagnos. De diagnostiska kriterierna uppdaterades för några år sedan och större vikt läggs nu än tidigare vid ögonsymtom [4].
Ehlers–Danlos syndrom av vaskulär typ. Ehlers–Danlos syndrom förekommer i flera olika former där vaskulär typ (typ IV) drabbar blodkärl. Syndromet orsakas av en mutation i COL3A1-genen, som kodar för typ III-kollagen som bidrar till kärlväggens hållfasthet. Syndromet kännetecknas av aneurysm och dissektion i medelstora artärer men även av tarmruptur. Bristningar i blodkärlen behöver inte föregås av aneurysm. Småledsöverrörlighet, genomskinlig hud och benägenhet för stora blåmärken är vanligt. Graviditetskomplikationer med artär- och uterusruptur är vanliga. Det är viktigt att känna till om en patient har denna sjukdom eftersom de mycket sköra blodkärlen kan komplicera operationer och ingrepp.
Allvarlig kärl- eller tarmhändelse inträffar ibland redan före 20 års ålder och oftast före 40 års ålder. Prevalensen av sjukdomen har uppskattats till 1/100 000 och hälften av de patienter som diagnostiseras har en nymutation. Män insjuknar vid lägre åldrar än kvinnor [5].
Loeys–Dietz syndrom. Loeys-Dietz syndrom (LDS), som först nyligen beskrivits, har det mest aggressiva förloppet av de kända ärftliga bindvävssyndromen. Detta tillstånd orsakas av mutationer i gener som kodar för TGFβ-signalkomplexet (TGFβR1, TGFβR2, TGFβ2 eller SMAD3). Hos majoriteten har mutationen uppstått de novo. Medellivslängden har beskrivits vara endast 26 år. Det är dock uppenbart att det finns familjer med mer godartade förlopp. Kärlruptur eller dissektioner sker vid mindre kärldiametrar än vid de övriga syndromen. Resultaten av förebyggande kirurgi är lyckligtvis goda. Syndromet är associerat med bifid eller bredbasig uvula, gomspalt och arteriell slingrighet som kan vara utbredd. Hypertelorism (ökat avstånd mellan de pariga organen, i regel ögonen), kraniosynostos, klubbfot, spondylolistes, mjuk och genomskinlig hud och benägenhet för blåmärken hör till den klassiska beskrivningen. Prevalensen anges till 1/1 000 000 men åtminstone i norra Sverige är prevalensen högre. Den höga morbiditeten som anges i litteraturen förklaras sannolikt av att syndromet är nyupptäckt och att få familjer med mildare symtom har hunnit diagnostiseras [6].
Andra sjukdomar associerade med TAAD
Bikuspid aortaklaff (BAV). Ca 1–2 procent av befolkningen har BAV. Av dem utvecklar 50–60 procent en vidgning av torakala aorta, och risken för aortadissektion är mångfaldigt ökad. Kirurgiskt ingrepp på klaffen och/eller aorta behövs hos ca 35 procent av patienterna.
Det finns en ärftlig komponent då 9 procent av förstagradssläktingar till en patient med BAV också har bikuspid aortaklaff. En gen av betydelse har hittills identifierats (NOTCH1). Alla utvecklar inte aortadilatation; olika grad av aortaengagemang har setts hos patienter med samma typ (fusion av samma kuspar) och samma grad av klaffdysfunktion. Den heterogena kliniska bilden orsakas sannolikt av en samverkan mellan genetiska faktorers inverkan på kärlstruktur och en förändrad hemodynamik i aorta [7].
Polycystisk njursjukdom. Polycystisk njursjukdom är förknippad med ökad risk för subaraknoidalblödning och aortadissektion [8]. Sjukdomen är en ärftlig multiorgansjukdom med cystor i båda njurarna, men även andra organ som lever och bukspottkörtel kan drabbas. Sekundär blodtrycksförhöjning kan komma tidigt medan njursvikten kan debutera sent. Prevalensen anges till 1/400–1 000 och anlaget är vanligtvis dominant nedärvt (95 procent). Syndromet orsakas av mutationer i PKD1- eller PKD2-genen.
Från mutation till sjukdom
Hittills har mutationer som orsak till icke-syndromatiska familjära TAAD identifierats i flera olika gener [9, 10]. Mutationer i ACTA2-genen anges vara den vanligaste förklaringen till ärftlig form av torakal aortadissektion och uppskattas utgöra ca 14 procent av fallen. Övriga gener är TGFβR1, TGFβR2, MYH11, SMAD3 och MYLK. Det finns även några kända lokus där sjukdomsgenerna ännu inte är identifierade. Familjära TAAD är genetiskt heterogena och det torde finnas flera okända gener som kommer att identifieras med tiden.
MYH11-, ACTA- och MYLK-generna kodar för proteiner i det kontraktila elementet i kärlens glatta muskelceller. Mutation i generna för dessa proteiner leder till en dysfunktion av muskelcellerna. De glatta muskelcellerna blir hyperplastiska vilket i sin tur ger upphov till en lägre elasticitet och aneurysmbildning i kärlväggen. Även apoptos av muskelceller kan ses.
Marfans syndrom orsakas av mutation i FBN1-genen som leder till en defekt i fibrillin. Fibrillin återfinns framför allt i proximala aorta. Mutation i proteinet leder till en minskad elasticitet i aorta ascendens och dessa patienter drabbas i regel av en vidgning av aortaroten.
Kollagen är ett viktigt stödjeprotein i kärlväggen och bidrar till kärlens hållfasthet. Den återfinns huvudsakligen längre distalt i kärlträdet vilket förklarar aneurysmbildningen och skörheten i mer perifera kärl vid Ehlers–Danlos syndrom av den vaskulära typen.
Uppreglering av TGF-β-signalering (transforming growth factor-beta) i aortaväggen ses vid Marfans syndrom, Loeys–Dietz syndrom samt familjära TAAD orsakade av MYH11 och ACTA2-mutationer [9]. Detta pekar på att TGF-β-signalering spelar en central roll vid aneurysmbildning vid dessa olika sjukdomar. TGF-β är en multifunktionell peptid som deltar i proliferation, differentiering och apoptos hos olika celler. Dess verkningsmekanism vid aneurysmbildning är ännu inte klarlagd.
I en dissekerad aorta kan histologiskt ses hyperplasi och apoptos av glatta muskelceller, degeneration av elastiska fibrer och en ökad förekomst av inflammatoriska mediatorer.
Varför utreda?
Efterfrågan på utredning från individer i de drabbade familjerna är stor. Anlagsbärare för familjära TAAD bör kontrolleras regelbundet och det finns förebyggande behandling att erbjuda. Den behandling som kan erbjudas i dag är:
Profylaktisk kirurgi. Profylaktisk kirurgi erbjuds i dag individer med aneurysm i aorta ascendens. Optimal tidpunkt för operation bör utgå från en avvägning mellan risk för dissektion och risk för operationskomplikationer. Sjukdomsgruppen är heterogen och dissektioner kan inträffa redan vid normala aortamått, varför rekommendationerna mest bygger på konsensus. Vid vilken diameter som man bör erbjuda patienten förebyggande operation varierar beroende på sjukdom och familjehistoria [11]. Som tumregel kan sägas att en ökning av aortadiametern på mer än 5 mm per år eller en diameter på 42–50 mm bör föranleda ställningstagande till operation. Det är framför allt vid dissektion i yngre ålder i familjeanamnesen och vid mutationer i vissa gener som man rekommenderar tidig operation.
Blodtrycksbehandling. Blodtrycksbehandling saknar evidens i form av kontrollerade studier men är mekanistiskt tilltalande. Med tanke på risken för dissektion rekommenderas därför ofta ett målblodtryck under 130/80 mm Hg.
Angiotensinreceptorblockerare (ARB). Allt fler data talar för att TGF (transforming growth factor)-systemet är överaktiverat vid många TAAD-syndrom. ARB blockerar TGF-β-receptorn och effekter av ARB på aneurysmutveckling har framför allt visats i djurmodeller [12]. Nyligen publicerades de första randomiserade studierna av losartan vid Marfans syndrom hos människa och de visade en långsammare aneurysmtillväxt än vid placebobehandling [13, 14]. I vilken omfattning detta enbart kan hänföras till medlets blodtryckssänkande effekt är ännu oklart.
Betablockerare. Betablockerare har använts i många år till denna patientgrupp och rekommenderas särskilt vid Marfans syndrom. Effekten av denna behandling vid Marfans syndrom hos vuxna har ifrågasatts i en systematisk översikt [15], och jämförande studier mot losartan pågår.
Med denna brist på kunskap är det svårt att ge entydiga riktlinjer om blodtrycksbehandling. Det är rimligt att behandla med både ARB och betablockad. Patienterna är dock ofta unga och har lågt blodtryck varför man ibland måste välja ett läkemedel. Vi väljer att på patientgrupper med känd aktivering av TGF-β-systemet (LDS, Marfans syndrom) prioritera ARB före betablockad, beroende på bättre kvalitet på de fåtal kliniska studier som finns. För patienter med Ehlers–Danlos syndrom av vaskulär typ väljer vi celiprolol [16].
Utredningsgång
Utredning vid misstänkta familjära TAAD kan utföras vid olika kliniker. Vi samarbetar inom ramen för CKG (Centrum för kardiovaskulär genetik) vid Norrlands universitetssjukhus i Umeå och har funnit det vara en effektiv organisationsmodell. Vid CKG arbetar kardiolog, barnkardiolog, internmedicinare och kliniska genetiker tillsammans. Två koordinatorer (biomedicinsk analytiker respektive sjuksköterska) håller samman arbetet och bedriver genetisk rådgivning.
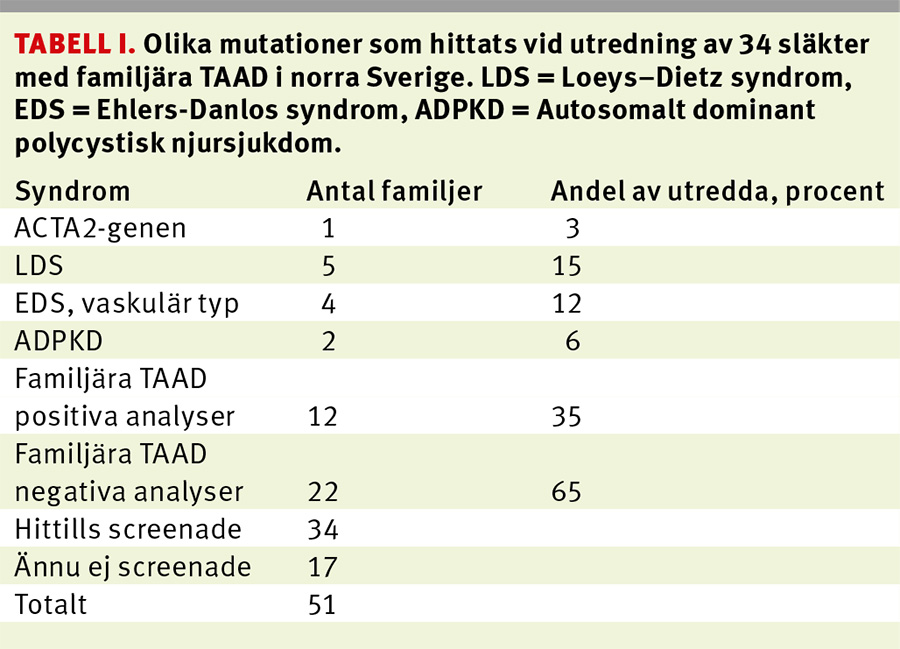
I december 2013 var 51 registrerade familjer under utredning vid CKG. För 34 av dessa fanns svar från den molekylärgenetiska utredningen (Tabell I).
De som remitteras för utredning är i första hand patienter med aortadissektion eller torakalt aortaaneurysm där det finns misstanke om familjär sjukdom. En annan grupp är patienter där man primärt misstänker ett bindvävssyndrom med aortaengagemang oavsett familjeanamnes.
Utredningens första steg är att rita ett släktträd och bekräfta att det finns en familjeanamnes. Detta moment kräver genomgång av släktens journalhandlingar, undersökningsresultat och obduktionsprotokoll. Den är vanligt att abdominella aortaaneurysm sammanblandats med torakala aortasjukdomar av såväl anhöriga som av sjukvården. Många av de ovanliga bindvävssyndromen har ofta förbisetts i sjukvården medan Marfans syndrom ofta har överdiagnostiserats.
Om familjeanamnesen kan verifieras eller det primärt finns misstanke om bindvävssyndrom kallas patienten för gemensam bedömning av klinisk genetiker och internmedicinare med inriktning mot kärlsjukdomar. Beroende på anamnes och fynd görs i nästa steg en molekylärgenetisk utredning av probanden eller kompletterande undersökningar, specifika för respektive syndrom.
Därefter bjuder vi in till en familjemottagning där alla i släkten som så önskar kan delta. Denna inleds med information om sjukdomen med åtföljande diskussion. Om den molekylärgenetiska analysen bekräftar diagnosen erbjuds probandens förstagradssläktingar anlagsbärartest. Om analysen inte påvisar någon mutation erbjuds första- och andragradssläktingar i stället en fenotypisk utredning med ekokardiografi, datortomografi eller magnetkamera.
Vi samarbetar för närvarande med andra laboratorier vid mutationsscreening för familjära TAAD. Kostnaden för en panel med 15 gener är 1 200 euro (2013). Om en sjukdomsorsakande mutation påvisas hos probanden erbjuds förstagradssläktingarna riktad mutationsanalys vilken utförs av Klinisk genetik, Norrlands universitetssjukhus.
Diskussion
I familjer där flera personer drabbats av aortadissektion finns ofta ett starkt önskemål om utredning, prevention och behandling. Såväl patienter med genomgången dissektion som deras släktingar har ofta haft svårt att få gehör för sin oro och sina frågor samt adekvat utredning och behandling i hälso- och sjukvården. Kunskapen om torakala aortasjukdomar är inte allmänt spridd inom vården. Ofta har en utredning begränsats endast till en ultraljudsundersökning av bukaorta.
Det finns inget svenskt nationellt handlingsprogram för familjära TAAD. Nordamerikanska riktlinjer rekommenderar att förstagradssläktingar till patienter med TAAD och identifierad mutation erbjuds provtagning för screening. För den majoritet av familjer där genanalyserna är negativa är rekommendationen att erbjuda fenotypisk screening till första- och andragradssläktingar [11].
Vi har försökt följa dessa riktlinjer, och själva utredningen är i många familjer tämligen lätt att genomföra. I andra familjer, särskilt de med hög debutålder, kan släkterna vara stora och många individer kan definieras som andragradssläktingar. Mycket få släktingar har avböjt utredning.
En praktisk svårighet är remissregler och debiteringssystem över landstingsgränserna. Många familjer har medlemmar som bor i andra landsting eller regioner och utredningen blir i vissa familjer uppsplittrad på många landsting och svår att överblicka.
Under arbetet med dessa TAAD-familjer har det blivit uppenbart att det finns stora kunskapsluckor kring dessa sjukdomar. I översiktsartiklar anges att man hittar den genetiska bakgrunden till sjukdomen hos endast 20 procent av familjerna, vilket avsevärt försvårar familjeutredningarna och senarelägger möjligheterna till specifik behandling. Den fenotypiska utredningen kan vara falskt negativ hos yngre individer, då det hos en del familjer inträffar aortadissektioner vid normala eller endast lätt vidgade aortamått. Vi har lyckats identifiera den bakomliggande orsaken hos 35 procent av familjerna, Ehlers–Danlos syndrom av vaskulär typ och polycystisk njursjukdom inkluderat (Tabell I).
Med denna bakgrund har vi ett forskningsprojekt där vi med exomsekvensering försöker identifiera nya gener av betydelse för TAAD. I projektet undersöks också om det finns andra sätt att påvisa fenotypen för familjära TAAD än att enbart mäta aortas diameter.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Torakalt aortaaneurysm och dissektion, TAAD
- TAAD kan utgöra en isolerad manifestation, vara en del i ett definierat syndrom eller en autosomalt dominant ärftlig sjukdom med nedsatt penetrans och variabel expressivitet. TAAD är genetiskt heterogen.
- Histologiskt ses degeneration av medialagret i aorta med förlust av glatta muskelceller, fragmentering av elastiska fibrer samt upplagring av muköst material.
- Marfans syndrom, Ehler–Danlos syndrom och Loeys–Dietz syndrom är de vanligaste syndromen som ger ökad benägenhet för TAAD.
- Den genetiska orsaken till ärftlig form av torakal aortadissektion har påvisats i cirka 20 procent av de familjära fallen. Prediktiv testning är möjlig i familjer där en sjukdomsorsakande mutation identifierats.
- Mutationer har identifierats i flera olika gener. De vanligaste är ACTA2, TGFBR1, TGFβR2, SMAD3, MYH11 och MYLK.
- Gener muterade vid familjära TAAD kodar för kontraktila delar av glatta muskelceller och proteiner i TGFβ-signalkomplexet.
Fakta 2. Utredningsgång vid nyupptäckt aneurysm/dissektion
- Bedöm förekomst av eventuellt bakomliggande syndrom utifrån kliniska kriterier för Marfans syndrom, Ehlers–Danlos syndrom och Loeys–Dietz syndrom.
- Utred familjär förekomst av torakal aortasjukdom.
- Om ingen familjeanamnes finns kan screening med avbildning av aorta övervägas hos förstagradssläktingar efter information om screeningens möjligheter och begränsningar.
- Vid misstanke om bindvävssyndrom eller verifierad familjeanamnes: gör en molekylärgenetisk analys i samråd med klinisk genetiker.
- Om familjeanamnes finns men genetisk analys är negativ: erbjud första- och andragradssläktingar screening med ekokardiografi, datortomografi eller magnetkamera efter information om sjukdomen.
- Alla individer med påvisad genmutation eller vidgade aortamått erbjuds kontroller för att värdera progress. Initiera blodtrycksbehandling.
- Om aortadiametern är normal görs individuell bedömning av förutsättningarna för anlagsbärarskap utifrån ålder och familjeanamnes.
Referenser
- Milewicz DM, Guo DC, Tran-Fadulu V, et al. Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annu Rev Genomics Hum Genet. 2008;9:283-302.
- Olsson C, Thelin S, Ståhle E, et al. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14 000 cases from 1987 to 2002. Circulation. 2006;114(24):2611-8.
- Coady MA, Davies RR, Roberts M, et al. Familial patterns of thoracic aortic aneurysms. Arch Surg. 1999;134(4):361-7.
- Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47(7):476-85.
- De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin Genet. 2012;82(1):1-11.
- Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275-81.
- Padang R, Bannon PG, Jeremy R, et al. The genetic and molecular basis of bicuspid aortic valve associated thoracic aortopathy: a link to phenotype heterogeneity. Ann Cardiothorac Surg. 2013;2(1):83-91.
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369(9569):1287-301.
- Pomianowski P, Elefteriades JA. The genetics and genomics of thoracic aortic disease. Ann Cardiothorac Surg. 2013;2(3):271-9.
- Gillis E, Van Laer L, Loeys BL. Genetics of thoracic aortic aneurysm: at the crossroad of transforming growth factor-beta signaling and vascular smooth muscle cell contractility. Circ Res. 2013;113(3):327-40.
- Hiratzka LF, Bakris GL, Beckman JA, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Circulation. 2010;121(13):e266-369.
- Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGF β signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358-61.
- Chiu HH, Wu MH, Wang JK, et al. Losartan added to beta-blockade therapy for aortic root dilation in Marfan syndrome: a randomized, open-label pilot study. Mayo Clin Proc. 2013;88(3):271-6.
- Groenink M, den Hartog AW, Franken R, et al. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: a randomized controlled trial. Eur Heart J. 2013;34(45):3491-500.
- Chun AS, Elefteriades JA, Mukherjee SK. Do blockers really work for prevention of aortic aneurysms? Aorta. 2013;1(1):45-51.
- Ong KT, Perdu J, De Backer J, et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet. 2010;376(9751):1476-84.
Summary
Thoracic aortic aneurysms and dissections (TAAD) can be divided into three different main categories.
1. Inherited syndromes predisposing to TAAD such as Marfan syndrome, Ehlers-Danlos syndrome type IV and Loeys-Dietz syndrome (less than 5% of all TAAD).
2. Familial TAAD (FTAAD) with more than one affected family member (20 % of all TAAD). Inheritance shows an autosomal dominant pattern and there are no features of known syndromes.
3. Sporadic forms of TAAD with no family history or features of syndromic forms.
FTAAD present earlier in life and dissections occur in smaller diameter than in sporadic cases. The underlying genetic cause can be found in about 20 % of the inherited cases. The pathogenesis seems to be an involvement of the transforming growth factor β (TGFβ) signaling pathway or a dysfunction of the smooth muscle cell contraction. The role of β-blockers for aneurysm prevention is uncertain and there are on-going studies comparing angiotensin receptor blockers and β-blockers.



















