Plötslig spädbarnsdöd definieras som oväntad spädbarnsdöd som inte kan förklaras av föregående anamnes, dödsplatsundersökning och obduktion.
Enligt trippelriskmodellen kan ett barn drabbas av plötslig spädbarnsdöd om det föreligger en kombination av kritisk ålder (särskilt 1–5 månader), medfödda riskfaktorer (passiv rökning) och yttre riskfaktorer (mag- eller sidoläge, samsovning).
Apnéer, autonom instabilitet och oförmåga till arousal har diskuterats som möjliga mekanismer, som också ifrågasatts.
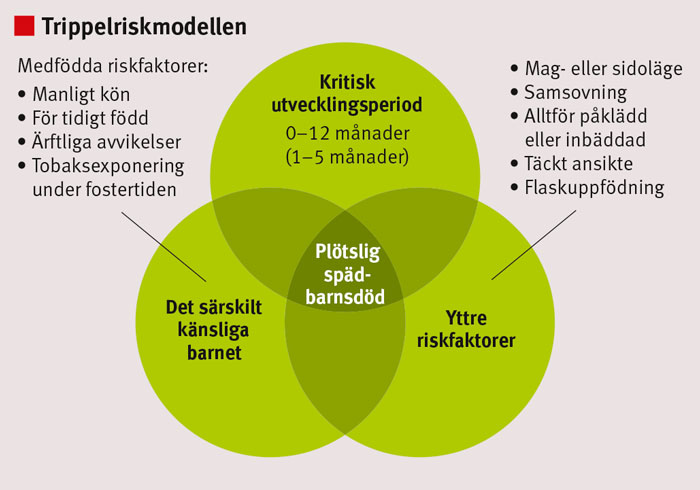
Figur 1. Trippelriskmodellen är den dominerande hypotesen om uppkomsten av plötslig spädbarnsdöd.
Plötslig spädbarnsdöd (SIDS; sudden infant death syndrome) har blivit föremål för en rad spekulationer. Vetenskapliga idéer om dess uppkomst slås upp stort i medier. De flesta av de hypoteser som lanserats har dock inte visat sig hålla. Antingen är de baserade på enstaka fall av plötslig spädbarnsdöd, som kanske inte är SIDS enligt definitionen, eller så är de helt enkelt felaktiga. De mest framträdande tänkbara patofysiologiska orsakerna till plötslig spädbarnsdöd diskuteras i denna översikt.
Forskningen om plötslig spädbarnsdöd har till stor del baserats på postmortala undersökningar (i samband med obduktion) och djurexperimentella modeller. Eftersom döden i princip är det första symtomet, har det varit omöjligt studera sjukdomsprocessen. Visserligen har s k livlöshetsattacker (ALTE; apparent life threatening events) undersökts ingående som tänkbara förstadier till plötslig spädbarnsdöd (near-SIDS; på svenska »nära plötslig spädbarnsdöd«), men det har också ifrågasatts.
Omfattande vetenskapliga undersökningar av barn som dött, barn som varit nära plötslig spädbarnsdöd och djurexperimentella modeller har inte resulterat i något genombrott som lett till en ordentlig minskning av fallen. Observationen att förekomsten av plötslig spädbarnsdöd minskar dramatiskt när spädbarn sover på rygg har haft en större betydelse.
Apnéhypotesen
Den dominerande hypotesen, särskilt under 1980-talet, rörande orsaken till plötslig spädbarnsdöd är att barn oväntat och plötsligt slutar andas (apné), vilket i sin tur leder till syrebrist och hjärtstopp (asystoli). Vidare observerades att s k riskbarn med avseende på plötslig spädbarnsdöd (barn med livlöshetsattacker, syskon till barn som dött och mycket för tidigt födda barn) hade fler apnéer än friska kontrollfall under sömn. Man diskuterade hur det kom sig att en sjukdom kunde debutera med döden och huruvida det inte borde finnas abortiva former, dvs barn som är på väg att dö i SIDS men räddas i sista stund. Begreppet »nära plötslig spädbarnsdöd« myntades.
Sambandet mellan apnéer och plötslig spädbarnsdöd blev föremål för en rad artiklar. I PubMed finner man inte mindre än drygt 1 150 referenser om man slår på »SIDS AND Apnea«. Stort intresse ägnades åt att studera mekanismer som kunde vara av betydelse för att hämma andningen. Denna forskning ansågs mycket relevant för att förklara plötslig spädbarnsdöd, eftersom dessa latenta hämningsmekanismer kunde finnas kvar efter födelsen och kanske utlösas under vissa betingelser.
Eftersom ett samband mellan apnéer och plötslig spädbarnsdöd kunde påvisas, blev det helt logiskt att övervaka särskilt riskbarn med apnémonitorer som larmar när barnet slutar andas mer än 20–30 sekunder. En rad olika typer av apnélarm har konstruerats genom åren, t ex rörelsekänsliga sensorer som fästs på bröstkorgen eller s k apnémadrasser. Detta har i USA utvecklats till en hel industri som omsätter hundratals miljoner dollar.
Redan på ett tidigt stadium ifrågasattes dessa larm. Apnéer visade sig vara ganska vanligt förekommande hos »friska« barn. I en omfattande studie fann man ingen skillnad i förekomst av s k extrema händelser mellan riskbarn (barn med livlöshetsattacker och syskon till barn som avlidit i plötslig spädbarnsdöd) och kontrollfall. Detta har konfirmerats i 11 studier, vilka sammanställts i en metaanalys [1].
Autonom obalans
Den norske fysiologen Birger Kaada lanserade därför en teori om att plötslig spädbarnsdöd beror på att ett spädbarn kan reagera med att bli förlamat av skräck och sluta andas och få pulsfall [2] – på norska »frukteparalys« eller på engelska »fear-paralysis reflex«. Denna teori väckte stor uppmärksamhet när den framlades år 1986. Kaada menade att reflexen kunde utlösas av att barnet plötsligt skrämts av föräldragräl och smällande dörrar [2]. Detta är ju en adekvat reaktion för att minska syrebehovet och överleva hos försvarslösa ungar som rådjurskid och harungar när de känner sig hotade av rovdjur. De spelar helt enkelt döda. Denna teori väckte upprörda känslor inte minst bland drabbade föräldrar, och några kliniska bevis presenterades aldrig, frånsett en del djurexperimentella observationer.
En liknande hypotes har framförts av Stephen Porges [3], som menar att en s k reptilvagus kan aktiveras vid stress hos spädbarn och leda till hjärtstopp (asystoli).
Flera olika metoder har utvecklats för att evaluera den autonoma balansen. En är spektralanalys av EKG, vilket innebär att man analyserar hjärtfrekvensvariabiliteten. Om pulsen registreras hjärtslag för hjärtslag visar det sig att den varierar beroende på vagus och sympatikus inverkan på hjärtats retledningssystem. Andningsrörelserna påverkar hjärtfrekvensen via vagus, den s k sinusarytmin, vilket ger upphov till en högfrekvent variabilitet. Stress stimulerar framför allt den lågfrekventa variabiliteten. Genom att bestämma kvoten mellan hög- och lågfrekvent variabilitet kan man få ett kvantitativt mått på den autonoma balansen. Avvikelser i hjärtfrekvensvariabilitet har upptäckts hos barn som dött i plötslig spädbarnsdöd [4].
Vagal aktivitet kan testas genom att trycka på ögonbulberna, snabb avkylning av pannan (cold face test), provokation av dykreflexen samt reaktion på plötslig ljudstimulering. Sambandet mellan vagal överreaktion och plötslig spädbarnsdöd har rapporterats endast i anekdotiska fallbeskrivningar, och mer systematiska undersökningar saknas [5] .
Ett mer skonsamt sätt att undersöka autonoma funktioner är att snabbt tippa barnet från liggande till stående läge [6]. Barnet får ligga i en specialgjord bädd med ett gångjärn, så att bädden snabbt kan lutas upp i 45–60 graders vinkel. När blodet då sjunker ner mot benen och blodtrycket därmed sjunker aktiveras tryckreceptorerna i halspulsådern, varvid vagus hämmas så att hjärtfrekvensen stiger och sympatikus aktiveras för att bibehålla blodtrycket. Denna mekanism är nödvändig för att vi inte ska svimma när vi hastigt reser oss upp.
Snabb tippning från liggande till stående har utförts i ett flertal studier för att identifiera riskbarn för plötslig spädbarnsdöd. För tidigt födda barn och bebisar till rökande mammor visade sig ha signifikant avvikande reaktioner när de tippades upp 60 grader. Intressant var att riskbarnen svarade med en blodtrycksreaktion, som kontrollbarnen, men att de sedan reagerade med blodtrycksfall. De verkade inte kunna finjustera det autonoma nervsystemet för att bibehålla den autonoma balansen (homeostasen).
Arousal och självåterupplivning
Eftersom plötslig spädbarnsdöd anses inträffa bara under sömn, har stort intresse ägnats åt spädbarnets förmåga att vakna upp, t ex om luftvägarna är tilltäppta. Vuxna snarkare med sömnapnéer vaknar till när de övre luftvägarna täpps till. Denna arousal-reaktion är ett slags kamp/flykt-reaktion när fara hotar, i detta fall syrebrist. Andningsrörelser, pulsfrekvens och blodtryck ökar för att avbryta apnén. Bebisar reagerar i mindre grad med arousal vid andningsstopp. I en studie omfattande 20 000 barn visade det sig att de barn som dog i plötslig spädbarnsdöd hade haft färre uppvaknanden, och de uppvaknanden som noterades verkade vara abnorma [7].
Genetisk orsak
Det finns en viss ökad risk för syskon till barn som dött i plötslig spädbarnsdöd att omkomma av samma orsak [8]. Tvillingar har visat sig ha ungefär dubbelt så hög risk att drabbas av plötslig spädbarnsdöd som »enlingar« (13 visavi 7 per 10 000 barn), men det fanns ingen skillnad mellan enäggs- och tvåäggstvillingar [9]. Plötslig spädbarnsdöd är dock vanligare i vissa etniska grupper, t ex indianer och afroamerikaner, men mer ovanligt bland barn med kinesiskt och japanskt ursprung som fötts i USA. Även om man korrigerar för socioekonomiska faktorer kvarstår dessa skillnader.
Jonkanalsjukdomar (QT-syndrom) är ärftliga och har upptäckts hos vissa barn som dött plötsligt. Det gäller även äldre barn och vuxna, ofta i samband med idrottsövningar. Detta tillstånd kan i princip upptäckas i förväg med EKG, varvid man funnit längre QT-intervall som tyder på en störning av hjärtats retledningssystem. I 5–10 procent av alla fall av plötslig spädbarnsdöd har det visat sig föreligga genförändringar associerade med QT-syndromet [10]. Huruvida dessa fall ska klassas som plötslig spädbarnsdöd kan diskuteras.
Avvikelser av gener som är av betydelse för utvecklingen av det autonoma nervsystemet, immunsystemet och metabolismen har rapporterats [11].
Kongenitalt centralt hypoventilationssyndrom har antagits ha ett visst samband med plötslig spädbarnsdöd [12]. Vid denna medfödda sjukdom kan barnet inte andas spontant när det sover, utan är då beroende av respirator, medan andningen kan vara normal i vaket tillstånd. Sjukdomen kallas också Undines förbannelse efter en medeltida myt om en sjöjungfru vars otrogna fästman straffades genom att bli fråntagen sin förmåga att andas automatiskt. Det är ett mycket ovanligt tillstånd och beror på en spontanmutation av PHOX2B-genen. Man har spekulerat om att vissa barn som antagits ha dött i plötslig spädbarnsdöd i själva verket haft kongenitalt centralt hypoventilationssyndrom.
Serotoninhypotesen
Av alla de neurotransmittorer som analyserats i hjärnan hos SIDS-offer är serotonin den som fått mest uppmärksamhet med 133 referenser i PubMed. Serotoninsystemet, eller 5-hydroxitryptamin (5-HT), är lokaliserat i hjärnstammens mittfåra (rafe) och anses vara involverat i sömnregleringen. Serotoninneuron har också upptäckts i de centrala kemoreceptorerna, som känner av koldioxidhalten i blodet och är av stor betydelse för andningskontrollen. Dessa receptorer är lokaliserade till hjärnstammens undersida vid fjärde ventrikeln och kan därmed reagera på vätejonförändringar, som i sin tur styrs av blodets koldioxidhalt. Serotoninsystemet projiceras till hjärnkärnor som reglerar andningen, blodtrycket, kroppstemperaturen, övre luftvägarna och förmågan att vakna upp (arousal), dvs funktioner vilkas bortfall kan leda till plötslig spädbarnsdöd.
Rubbning av serotoninsystemet som möjlig orsak till plötslig spädbarnsdöd har rapporterats från framför allt Hannah Kinneys forskargrupp i Boston. I en mycket uppmärksammad studie visade de att antalet serotoninreceptorer var signifikant färre i flera hjärnkärnor som är engagerade i sömn- och andningskontroll [13]. Detta noterades särskilt hos pojkar som dött i plötslig spädbarnsdöd. Å andra sidan fann de ökat antal serotonininnehållande nervceller än hos kontroller. Deras tolkning av dessa lite paradoxala fynd är att det ökande antalet serotoninneuron som sågs vid SIDS har lett till en alltför hög extracellulär serotoninnivå, vilket sekundärt resulterat i en nedreglering av antalet serotoninreceptorer. Rapporten är baserad på en postmortal studie av 31 SIDS-fall och 10 kontroller. Tio av dessa indexfall var för tidigt födda barn, varför man inte kan utesluta att fyndet beror på skillnader i utvecklingsgrad. Dessutom förekom flera riskfaktorer hos barnen som avlidit i SIDS, t ex magläge och nikotinexponering.
I en ny studie från Boston framfördes ytterligare bevis för serotoninhypotesen. Denna studie är baserad på 41 SIDS-fall, 7 kontroller som dött av helt andra orsaker och 5 barn som legat på sjukhus och avlidit i kronisk syrebrist. Serotoninhalten visade sig vara signifikant lägre i flera hjärnkärnor hos SIDS-offren [14], vilket motsäger det tidigare fyndet av fler serotoninnervceller vid SIDS. Forskargruppen i Boston kan inte förklara denna diskrepans, eftersom kliniska data för index- och kontrollgrupperna är likartade. Däremot konfirmerades fyndet av lägre receptorbindning hos tre hjärnkärnor i hjärnstammen, som mottar serotonerga projektioner hos SIDS-gruppen än hos kontrollgruppen. Den tredje gruppen av barn (barn som dött av svår syrebrist) hade ungefär samma halter av serotoninmarkörer som övriga kontroller. Detta tolkas som att avvikelserna av serotoninnivåer och receptorer inte beror på kronisk hypoxi.
Serotoninhypotesen stärks av fyndet av två funktionella genpolymorfismer av serotonintransportgenen, som visade sig vara vanligare hos SIDS-offer än hos kontroller [15]. Dessa fynd har kunnat konfirmeras i en studie, medan andra studier har givit negativa resultat [11]. Diskrepansen kan förklaras av att dessa genpolymorfismer huvudsakligen påträffas hos afroamerikaner men inte hos barn med europeiskt ursprung.
Noradrenalin
Noradrenalinet är det sympatiska nervsystemets viktigaste neurotransmittor och därmed av betydelse för att upprätthålla blodtryck och hjärtfrekvens, särskilt vid stress. Nikotin har i en rad studier visat sig rubba noradrenalinsyntes och -frisättning, vilket kan vara en av förklaringarna till att rökning ökar risken för plötslig spädbarnsdöd [16].
Det noradrenerga systemet i hjärnan är av stor betydelse för uppvaknande (arousal), uppmärksamhet och andnings- och blodtrycksreglering. Noradrenerga nervceller är till stor del lokaliserade i locus caeruleus, varifrån nerver utgår till hjärnbarken och hjärnkärnor i hjärnstammen som är engagerade i den homeostatiska kontrollen.
Eftersom defekt arousal påvisats hos barn som dött i plötslig spädbarnsdöd, har det varit naturligt att misstänka att det noradrenerga systemet är omoget eller defekt hos dessa barn [17]. Någon relation mellan genförändringar av två andra enzym involverade i noradrenalinomsättningen och plötslig spädbarnsdöd har dock ej kunnat påvisas [18].
Acetylkolin
Stort intresse har ägnats åt acetylkolin och plötslig spädbarnsdöd. Acetylkolin är inte bara vagussystemets neurotransmittor, utan också av betydelse för hjärnans medvetandegrad och andningskontroll. Acetylkolin utövar sina effekter via muskarin- och nikotinreceptorer. De senare påverkas av tobaksexponering, en väl etablerad riskfaktor för plötslig spädbarnsdöd. Möss som exponerats för nikotin i fosterlivet visade sig ha nedsatt arousal-respons. Samma effekt noterades hos knockoutmöss där de β-2-nikotinerga acetylkolinreceptorerna slagits ut [16]. Det är möjligt att nikotinexponering under fosterlivet påverkar acetylkolinreceptorernas känslighet (desensitisering) [16]. Kliniska studier har visat att nikotinexponering under fosterlivet och spädbarnstiden försämrar homeostasmekanismer hos spädbarnet [6, 19].
Trippelriskmodellen
Den dominerande hypotesen om uppkomsten av plötslig spädbarnsdöd är den s k trippelriskmodellen [20] (Figur 1). Det antas att det finns en underliggande genetisk faktor, såsom en rubbning av serotoninsystemet. Nikotinexponering under fostertiden kan också störa neurotransmittorbalansen. När barnet är 2–4 månader gammalt befinner det sig i en särskild riskperiod, kanske beroende på autonom instabilitet. Buk- eller sidoläge kan utlösa ett pulsfall, som leder till sänkt syremättnad. Om barnet inte vaknar till genom arousal eller mobiliserar andra försvarsåtgärder, kan det dö i plötslig spädbarnsdöd.
En invändning mot denna modell är att risken borde vara mera uttalad ju yngre barnet är. Å andra sidan är vagussystemet, som bromsar hjärtaktiviteten, mer moget från och med 2 månaders ålder, medan det nyfödda barnet har större möjligheter att mobilisera det sympatikoadrenala systemet vid stress. Det nyfödda barnet är till och med utrustat med extra adrenalindepåer i form av paraganglier, som försvinner efter några månader.
Varför bukläge ökar risken är fortfarande helt oklart. Det verkar inte påverka den autonoma balansen vid provokation. En tänkbar förklaring är att barnets värmereglering påverkas. Ansiktet är mycket viktigt för spädbarnets värmeavgivning och om det ligger mot madrassen försvåras denna. Särskilt när barnet blivit några månader gammalt och lagt på hullet, är det viktigt att ansiktet är fritt. En temperaturhöjning, särskilt av hudtemperaturen, kan tänkas utlösa apné och pulsfall.
Fungerar hos över 99,9 procent av alla spädbarn
Hittills har ingen direkt orsak till plötslig spädbarnsdöd upptäckts. Frågan är om plötsliga dödsfall hos barn med vissa genetiska avvikelser verkligen ska rubriceras som plötslig spädbarnsdöd. Observationen att monozygota inte drabbas oftare än dizygota tvillingar talar emot en genetisk orsak.
Däremot har man identifierat en rad olika faktorer, som extremt för tidig födsel och nikotinexponering samt – framför allt – sovläge på magen, som riskfaktorer. Vidare har man identifierat konstitutionella faktorer, dvs att vissa homeostatiska mekanismer är omogna hos alla spädbarn, särskilt mellan 1 och 4 månaders ålder. Det märkliga kanske är att dessa mekanismer fungerar så bra hos >99,9 procent av alla spädbarn. Mekanismerna för att andning och hjärtverksamhet förblir kontinuerlig under olika omständigheter och inte rubbas irreversibelt är också ett mysterium.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Referenser
- Strehle EM, Gray WK, Gopisetti S, et al. Can home monitoring reduce mortality in infants at increased risk of sudden infant death syndrome? A systematic review. Acta Paediatr. 2012;101:8-13.
- Kaada B. Hjertedöd utlöst ved »frukteparalyserefleksen«. Tidsskr Nor Laegeforen. 1986;106:894-8.
- Porges SW. Orienting in a defensive world: mammalian modifications of our evolutionary heritage. A Polyvagal Theory. Psychophysiology. 1995;32:301-18.
- Schechtman VL, Raetz SL, Harper RK, et al. Dynamic analysis of cardiac R-R intervals in normal infants and in infants who subsequently succumbed to the sudden infant death syndrome. Pediatr Res. 1992;6:606-12.
- Wennergren G, Hertzberg T, Milerad J, et al. Hypoxia reinforces laryngeal reflex bradycardia in infants. Acta Paediatr Scand. 1989;78(1):11-7.
- Cohen G, Vella S, Jeffery H, et al. Cardiovascular stress hyperreactivity in babies of smokers and in babies born preterm. Circulation. 2008;118(18):1848-53.
- Kato I, Franco P, Groswasser J, et al. Incomplete arousal processes in infants who were victims of sudden death. Am J Respir Crit Care. Med. 2003;168:1298-303.
- Hunt CE. Genes and sudden infant death syndrome. Pediatr Res. 2004;56:321-2.
- Mitchell EA, Elder DE, Zuccollo J. Simultaneous sudden unexpected death in infance of twins: case report. Int J Legal Med. 2010;124:631-5.
- Arnestad M, Crotti L, Rognum TO, et al. Prevalence of long QT-syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361-7.
- Van Norstrand DW, Ackerman MJ. Genomic risk factors in sudden infant death syndrome. Genome Med. 2010;2:86.
- Rand CM, Patwari PP, Carroll MS, et al. Congenital central hypoventilation syndrome and sudden infant death syndrome: Disorders of autonomic regulation. Semin Pediatr Neurol. 2013;20:44-55.
- Paterson DS, Trachtenberg FL, Thompson EG, et al. Multiple serotonergic brainstem abnormalities in sudden infant death syndrome. JAMA. 2006;296:2124-32.
- Duncan JR, Paterson DS, Hoffman JM, et al. Brainstem serotonergic deficiency in sudden infant death syndrome. JAMA. 2010;303:430-7.
- Weese-Mayer DE, Berry-Kravis EM, Maher BS, et al. Sudden infant death syndrome: association with a promotor polymorphism of the serotonin transmporter gene. Am J Hum Genet. 2003;117A:268-74.
- Cohen G, Roux JC, Grailhe R, et al. Perinatal exposure to nicotine causes deficits associated with a loss of nicotinic receptor function. Proc Natl Acad Sci U S A. 2005;102(10):3817-21.
- Ozawa Y, Takashima S. Developmental neurotransmitter pathology in the brain stem of sudden infant death syndrome: a review and sleep position. Forensic Sci Int. 2002;130S:S53-9.
- Klintschar M, Heimbold C. No association of SIDS with two polymorphisms in genes relevant for the noradrenergic system. Acta Paediatr. 2012;101:1079-82.
- Cohen G, Jeffery H, Lagercrantz H, et al. Long-term reprogramming of cardiovascular function in infants of active smokers. Hypertension. 2010;55(3):722-8.
- Trachtenberg F, Haas EA, Kinney, HC, et al. Risk factor changes for sudden infant death syndrome after initiation of Back-to Sleep campaign. Pediatrics. 2012;129:630-8.
I Barnpuder ingår talk som har medicinska risker:
http://m.medlineplus.gov/ency/article/002719.htm
Citat - http://m.medlineplus.gov/ency/article/002719.htm?page=3 -
"Symptoms
Most symptoms are caused by accidental or long-term breathing in (inhaling) of talc dust, especially in infants. Breathing problems are the most common problem."
Andra problem:
* Heart and blood
* Collapse
* Convulsions
* Low blood pressure
* Lungs
* Chest pain
* Cough
* Difficulty breathing
* Lung failure
* Rapid, shallow breathing
* Nervous system
* Coma
* Drowsiness
* Fever
* Lack of desire to do anything (lethargy)
* Twitching of arms, hands, legs, or feet
* Twitching of the facial muscles
Fler tips på MedlinePLUS:
If the person breathed in the talcum powder, immediately move him or her to fresh air.
Breathing in talcum powder can lead to very serious lung problems, even death
Use caution when using talcum powder on babies. Talc-free baby powder products are available.
Serious lung damage and cancer have also been reported in workers who have breathed in talcum powder many times over long periods of time.
Intravenous use of street heroin that contains talc may lead to heart and lung infections and serious organ damage and death.
Alternative Names
Talc poisoning; Baby powder poisoning
Jag gjorde många hembesök som allmänläkare och kunde ibland notera hur mammorna pudrade en del bebisar försiktigt kring halsen, andra skapade vita moln runt ansiktet på barnet. Jag kunde varken då eller nu finna några referenser/undersökningar om förekomst av talk i luftvägar vid SIDS så jag ringde rättsmedicinen i Lund och framförde mina observationer till Gerhard Voigt. Han blev intresserad och hade möjligheter att specialfärga preparat från SIDS-fall han sparat, men han rapporterade muntligen till mig att han inte kunde påvisa talk i dessa. Han frågade om jag hade fler idéer - bara om infektioner svarade jag. Infektion förklarar ju nyttan med ryggläge, rökfrihet och ökningen under den kalla årstiden, och att SIDS är vanligare i sk socialgrupp 3. Aerosoler (inandning) kräver mycket lägre smittdos. Jag informerade dammsugartillverkare om detta - start via Allergiutredningen då. HEPA-filter började sedan kunna köpas.
Hittar en intressant artikel när jag omigen söker på SIDS - "A unifying theory"
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2798085/
Den förtjänar att vi läser den.