




Familjär medelhavsfeber är i typfallet en autosomalt recessiv sjukdom som karakteriseras av återkommande självbegränsande feberattacker med en duration på 12–72 timmar.
Feberattackerna är förenade med peritonit, pleurit och/eller artrit.
Majoriteten av patienterna har sin sjukdomsdebut under barndomen.
Sjukdomen bör misstänkas framför allt hos individer med ursprung i östra Medelhavsområdet.
Kolkicin ger effektiv behandling. Utan behandling är risken att utveckla amyloidos betydande.
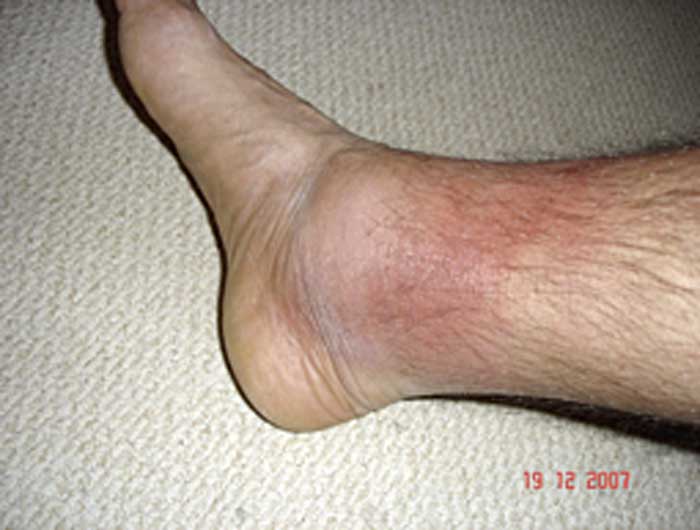
Figur 1. Hudutslag vid familjär medelhavsfeber: erysipelasliknade utslag med typisk lokalisation i samband med en feberepisod.
Foto: Helen Lachmann
Familjär medelhavsfeber tillhör gruppen autoinflammatoriska sjukdomar, vilka karakteriseras av ofta livslångt återkommande episoder av inflammation kopplade till feber i kombination med symtom såsom buksmärtor, trötthet, ledvärk och hudutslag. De autoinflammatoriska sjukdomarna är huvudsakligen medierade av celler och molekyler inom det medfödda immunsystemet med en mer eller mindre tydlig ärftlighet [1]. Familjär medelhavsfeber är i typfallet en autosomalt recessiv sjukdom som kännetecknas av återkommande feberattacker med en duration på 12–72 timmar förenade med serosit, vanligtvis peritonit, men också pleurit och/eller artrit. Utan behandling är sjukdomen förknippad med en betydande risk att utveckla amyloidos (Fakta 1).
På 1970-talet visades att kolkicin är en effektiv behandling, men innan dess var familjär medelhavsfeber en sjukdom med betydande morbiditet och mortalitet [2, 3] och orsakade avsevärt lidande med plågsamma attacker, amyloidos och för tidig död i njursvikt hos många drabbade patienter. Introduktionen av kolkicin innebar att situationen förändrades totalt för denna patientgrupp. I dag är medellivslängden jämförbar med befolkningens i övrigt, och risken för amyloidosutveckling är i princip begränsad till patienter utan eller med bristande följsamhet till behandling [4, 5].
För några år sedan visade vi att prevalensen i Västra Götalandsregionen bland svenska invånare med ursprung i östra Medelhavsområdet var i samma storleksordning som i ursprungslandet. Prevalensen hos invånare av turkiskt ursprung var 173 per 100 000 individer, av libanesiskt ursprung 124 per 100 000 och av syriskt ursprung 86 per 100 000 [6].
Med den demografiska utveckling Sverige genomgått sedan andra världskrigets slut är det uppenbart viktigt att identifiera, diagnostisera och behandla patienter med familjär medelhavsfeber i Sverige i dag. Speciellt angeläget är det inom barn- och ungdomssjukvården, eftersom nästan alla dessa patienter har sina första symtom före 18 års ålder.
Syftet med denna artikel är att bidra till ökad medvetenhet och förbättrad kunskap om sjukdomen genom att diskutera epidemiologi, sjukdomsmekanismer, ärftlighet, klinisk bild, diagnostik och behandling.
Epidemiologi
Familjär medelshavsfeber är den vanligaste monogent ärftliga autoinflammatoriska sjukdomen i världen med uppskattningsvis drygt 100 000 drabbade individer [7, 8]. Den är särskilt vanlig hos personer med ursprung i östra Medelhavsområdet, såsom turkar, araber, armenier och israeler. I dessa befolkningar är prevalensen mellan 100 och 200 per 100 000 individer, och i vissa områden är 20 procent av befolkningen bärare av en mutation i genen MEFV (Mediterranean fever) [8].
I Turkiet är prevalensen 100–200 per 100 000 invånare beroende på i vilken del av landet som undersökningen genomförts, med ännu högre prevalens i vissa delar av landet [9]. I Libanon och Syrien är prevalensen okänd, vilket även är fallet för arabiska länder i allmänhet [7]. Armenien har sannolikt den högsta prevalensen, uppskattad till 200 per 100 000 invånare [7]. Så vitt vi vet är förekomsten i Irak inte känd.
Det har hävdats att den enda möjliga förklaringen till den höga frekvensen bärare av MEFV-mutationer är att individer med en mutation har haft en evolutionär överlevnadsfördel i östra Medelhavsområdet [10], såsom ökad motståndskraft mot någon infektion. Sjukdomen är dock inte begränsad till befolkningar i östra Medelhavsområdet, utan ett ökande antal patienter diagnostiseras bland personer med ursprung i andra delar av världen, t ex Grekland, Italien, Japan, Indien, Kina och Storbritannien [8, 10].
Ärftlighet
Genen som är associerad med familjär medelhavsfeber, MEFV, identifierades 1997 på kromosom 16p13 och kodar för proteinet pyrin [11, 12]. De två forskargrupper som identifierade MEFV beskrev fyra mutationer: M680I, M694V, V726A och M694I. Dessa är fortfarande de vanligaste mutationerna i befolkningar med hög förekomst av sjukdomen [13]. I dag har ca 300 olika MEFV-mutationer identifierats, varav 80–90 är associerade med klinisk sjukdom, medan de återstående 200 varianterna antingen är av okänd betydelse eller betraktas som polymorfismer, exempelvis E148Q och P369S [14].
Familjär medelshavsfeber betraktas i typfallet som en autosomalt recessiv sjukdom. När patienter med kliniskt diagnostiserad sjukdom screenas för de 10–12 vanligaste mutationerna är ungefär 75 procent homozygota eller sammansatt heterozygota, dvs de har två sjukdomsframkallande mutationer som förväntat [6, 15]. Emellertid påvisas antingen en enda mutation eller ingen mutation alls hos en betydande andel (ungefär 25 procent) av patienter med klinisk sjukdom (se fallet Sabina, Fakta 1) [8].
Den enklaste förklaringen till detta är att screeningen av vanliga mutationer förbiser mindre vanliga, men likväl sjukdomsframkallande, mutationer. Denna förklaringshypotes har dock inte kunnat bekräftas med sekvensering av hela MEFV-genen och dess promotorregion [16]. En annan förklaringsmodell är att somatisk mosaikism bidrar till sjukdomsuttrycket hos patienter med endast en mutation (heterozygota patienter). Denna mekanism har nyligen bidragit till att förklara den stora andelen mutationsnegativa patienter som återfinns inom flera autosomalt dominanta autoinflammatoriska sjukdomar, men har inte ännu studerats vid familjär medelhavsfeber [17].
Det har även föreslagits att mutationer i gener uppströms eller nedströms MEFV skulle kunna bidra till sjukdomen hos heterozygota och mutationsnegativa patienter [18].
Ytterligare en förklaring som framhållits är att familjär medelshavsfeber inte alltid är en autosomalt recessiv sjukdom utan att en autosomalt dominant form är vanligare än man tidigare trott. Att det förekommer autosomalt dominant nedärvd familjär medelshavsfeber har visats hos bl a ett par familjer med brittiskt ursprung, där klinisk sjukdom är kopplad till ett autosomalt dominant ärftlighetsmönster [10].
Sammanfattningsvis är ärftligheten vid sjukdomen i många stycken komplex och i vissa stycken svårförståelig, vilket gör att god kompetens och stor erfarenhet är nödvändig för adekvat tolkning av genetiska undersökningsresultat.
Sjukdomsmekanismer
När genen för familjär medelshavsfeber identifierades för nästan 20 år sedan var förväntningarna stora på att man även skulle kunna förklara pyrins molekylära och patofysiologiska funktion [11, 12]. Dessa förväntningar har inte infriats, utan proteinets funktion är fortfarande oklar [17]. Den gängse uppfattningen har länge varit att pyrin normalt inhiberar ett immunreglerande proteinkomplex, den s k NLRP3-inflammasomen, vars aktivitet leder till produktion av interleukin 1-beta (IL-1β).
Enligt hypotesen att MEFV-mutationer hos individer med familjär medelhavsfeber ger upphov till en »loss-of-function«-mutation (dvs en mutation som leder till minskad funktion) hos pyrin hämmas NLRP3-inflammasomen mindre effektivt, vilket i sin tur leder till ökad produktion av IL-1β och därmed ökad inflammation [19]. Hypotesen att pyrin samverkar med NLRP3 har ifrågasatts baserat på studier, både på mus och på människa, vilka i stället talar för att pyrin bildar en egen inflammasom oberoende av NLRP3 [20].
Vidare visar studier på möss att vissa MEFV-mutationer i själva verket ger upphov till en »gain-of-function«-mutation (mutation som leder till ökad funktion) som i sig resulterar i ökad IL-1β-sekretion [20], vilket helt motsäger hypotesen ovan. Även om denna publikation är elegant har motsvarande mekanismer inte undersökts hos människa.
Det är mycket möjligt att pyrin påverkar inflammationsregleringen även via andra mekanismer än reglering av IL-1β-produktion, företrädesvis via aktivering av den proinflammatoriska transkriptionsfaktorn NF-κB [10]. En färsk studie tyder på att pyrin aktiveras genom att känna av att det kroppsegna enzymet rho-GTPas när detta modifierats av bakteriella toxiner från Clostridium difficile, Vibrio parahaemolyticus och Clostridium botulinum [21]. Detta skulle kunna ge ledtrådar till den evolutionära överlevnadsfördel som bärarskap av mutation i MEFV tycks medföra [10, 22].
Sammanfattningsvis är det, trots att det har gått snart 20 år sedan upptäckten att mutationer i MEFV leder till familjär medelhavsfeber, fortfarande i stora drag oklart hur detta sker. Möjligen kan den nya upptäckten att pyrin aktiveras av bakteriellt modifierat GTPas bidra till ökad förståelse av sjukdomsmekanismerna.
Klinisk bild
Familjär medelhavsfeber karakteriseras av återkommande feberattacker med en duration på 12–72 timmar förenade med buksmärtor (peritonit), bröstsmärtor (pleurit) eller artrit (synovit). Det vanligaste symtomet är buksmärtor, ofta kraftiga, orsakade av peritonit.
Den kliniska bilden är svår att skilja från blindtarmsinflammation, och en betydande andel av patienterna är laparotomerade innan de diagnostiseras [23]. Den pleuritiska bröstsmärtan är nästan alltid ensidig, och smärtan under en attack kan börja i bröstet och sedan röra sig ner till buken [2]. Associerad artrit pågår ofta längre än själva feberepisoden och engagerar främst stora leder i de nedre extremiteterna (höfter, knän och anklar); de misstas ofta för septisk artrit. Perikardit och orkit är ovanliga manifestationer under attackerna, men förekommer.
Ett typiskt kännetecken för sjukdomen är ett erysipelasliknande erytem med skarpt avgränsat ömmande plack, ofta på fotryggen, underbenet eller ankelområdet (Figur 1).
Hos barn varierar buksmärtornas intensitet, från lindriga övergående attacker till fulminant peritonit. Inflammationen hämmar ofta peristaltiken, vilket leder till förstoppning [2, 24]. Under 2 års ålder kan feberepisoder vara den enda sjukdomsmanifestationen, vilket understryker svårigheten att diagnostisera och utesluta familjär medelhavsfeber hos de minsta barnen [25]. I denna situation är genetisk analys ofta det enda sättet att föra diagnostiken i bevis.
Under sjukdomsattackerna påverkas de inflammatoriska markörerna med en ökning av neutrofila granulocyter, CRP och S-amyloid A (SAA). SAA har visat sig vara en känslig markör för inflammation vid autoinflammatoriska sjukdomar och produceras i levern efter stimulering med proinflammatoriska cytokiner. SAA är inte specifik för autoinflammation, eftersom den ofta är förhöjd vid andra inflammatoriska och infektiösa sjukdomar. Under en sjukdomsattack är SAA-värdet ofta >300 mg/l, den övre detektionsnivån på vårt laboratorium. Vid en sjukdomsattack förknippad med tydlig CRP-stegring adderar inte ett förhöjt SAA-värde >300 mg/l någon väsentlig information. Däremot kan analys av SAA hjälpa till att skärpa diagnostiken hos individer utan tydlig CRP-stegring under attackerna.
Subklinisk inflammation är vanligt mellan attacker hos patienter utan behandling, men kan också vara uttryck för bristande följsamhet till behandlingen och för låg dos av kolkicin hos behandlade patienter [26]. Den allvarligaste komplikationen till familjär medelhavsfeber är AA-amyloidos, som oftast är lokaliserad till njurarna, med proteinuri som ett viktigt och tidigt kliniskt tecken. På sikt är njur-AA-amyloidos förknippad med betydande risk för njursvikt. SAA är en känslig markör för subklinisk inflammation vid familjär medelhavsfeber, och eftersom proteinet är ett förstadium till amyloid A, som inlagras vid AA-amyloidos, är det möjligt att följa behandlingseffekten och värdera risken för amyloidos med mätning av SAA. I sällsynta fall kan även symtomfria patienter utan attacker utveckla amyloidos, sannolikt orsakat av subklinisk inflammation [2, 27].
Barn med familjär medelhavsfeber mår oftast bra mellan attackerna, men ansträngningsutlöst smärta i benen är förhållandevis vanlig och förebyggs inte av kolkicin, men kan behandlas med NSAID [27]. Barn med sjukdomen kan även utveckla febril myalgi som yttrar sig som subfebrilitet och muskelsmärtor under flera veckor utanför de typiska attackerna. Tillståndet orsakas sannolikt av vaskulit och behandlas med steroider [27].
Diagnostik
Familjär medelhavsfeber är i många stycken en klinisk diagnos, vilket understryks av att den genetiska analysen i dagsläget kan bekräfta men inte utesluta sjukdomen [8]. År 1997 formulerades de förenklade Tel Hashomer-kriterierna, vilka framgår av Fakta 2 [28]. Nästan alla patienter har sjukdomsdebut under barnaåren, och diagnoskriterier för barn föreslogs 2009 (Fakta 3) [29]. Dessa kriterier kan vara till hjälp vid diagnos, men sensitivitet och specificitet är avhängiga prevalensen av sjukdomen i den aktuella befolkningen [8], och både läkare och föräldrar vill ofta söka stöd för en klinisk diagnos eller misstanke i en genetisk undersökning.
En klinisk diagnos är dock inte alltid möjlig, vilket exemplifieras av de allra yngsta barnen, där feberepisoderna kan vara det enda tecknet på sjukdomen [25]. När den genetiska utredningen inte bekräftar den kliniska misstanken, får man återvända till den kliniska bilden och utvärdera den igen, ofta tillsammans med resultatet av behandlingsförsök med kolkicin [8].
Behandling
Kolkicin är en effektiv behandling och förändrar ofta dramatiskt livet för drabbade patienter [3]. Före kolkicinerans början på 1970-talet utvecklade patienter vanligtvis amyloidos före 40 års ålder, som följd av förhöjd nivå av SAA sekundärt till återkommande attacker och subklinisk inflammation [2]. I ett första steg syftar behandlingen till att förhindra attacker och i ett andra till att normalisera inflammationen mellan episoderna och därmed förhindra utveckling av amyloidos, för vilket serumnivåerna av SAA är en känslig markör [26]. Då patienten inte svarar på kolkicin måste bristande följsamhet eller för låg behandlingsdos övervägas [7, 8].
Det är inte känt vilken nivå av SAA mellan episoder som inte är förknippad med risk för utveckling av amyloidos, men kolkicinbehandling ger oftast fullgott skydd [4, 5]. I det kliniska arbetet är vi nöjda om SAA är <20 mg/l hos patienter utan amyloidos, men får ofta acceptera ett värde mellan 20 och 30 mg/l. Förutom avsaknad av behandling och bristande följsamhet till behandling är risken för att utveckla amyloidos kopplad till vissa mutationer i MEFV, framför allt M694V, men även till vistelseland och SAA1 α/α-genotyp [7, 8].
Vid insättning av kolkicin kan patienten utveckla övergående känslighet för laktos, vilken behandlas med reduktion av laktosintaget. Gastrointestinala biverkningar av kolkicin kan göra det svårt att nå den önskade dosen; då kan det vara en fördel att ge dygnsdosen uppdelad på två tillfällen i stället för ett, vilket är den normala doseringen.
Kolkicin finns i kapsel på 0,25 mg (ex tempore-beredning, Apotek Produktion & Laboratorier AB [APL]) och en nyligen godkänd tablett (Colrefuz) på 0,5 mg. Även om det är ovanligt att patienter inte svarar på kolkicin eller inte tolererar medicineringen, förekommer det. Dessa patienter behandlas vanligtvis effektivt med IL-1-blockad [8]. Det finns en liten risk att patienter som behandlas med kolkicin utvecklar vitamin B12-brist, vilket gör att B12 bör kontrolleras inför behandlingsstart och regelbundet under behandlingen.
Diskussion
Familjär medelhavsfeber har beskrivits hos befolkningar i stora delar av världen, men är vanligast hos personer med ursprung i östra Medelhavsområdet. Efter andra världskriget har Sverige haft en betydande invandring från länder med hög förekomst av sjukdomen, såsom Turkiet och arabiska länder [30]. Detta har gjort att sjukdomen, som tidigare var mycket ovanlig i Sverige, blivit allt vanligare bland svenska invånare i dag. Sjukdomen är förknippad med svåra självbegränsande feberattacker under en halv till 3 dagar, kopplade till buksmärtor, bröstsmärtor (pleurit) eller artrit (synovit), men också med en betydande risk för utveckling av amyloidos.
Är man som läkare medveten om diagnosen är sjukdomsbilden vanligtvis inte svår att känna igen, och behandling med kolkicin förhindrar inte bara attackerna utan förebygger även amyloidos hos i princip alla patienter.
Amyloidos, företrädesvis njuramyloidos, är fortfarande den viktigaste dödsorsaken hos patienter med familjär medelhavsfeber, trots att kolkicin drastiskt minskat risken för amyloidosutveckling. Proteinuri är ett tidigt tecken på njuramyloidos och fångas med vanlig urinsticka. I stort sett är utveckling av amyloidos begränsad till obehandlade och behandlade patienter med bristande följsamhet [4, 5, 7]. I Turkiet har risken för att utveckla amyloidos minskat från 60 procent i en studie från 1996 till <13 procent i en studie från 2005 [31, 32].
I dagens globaliserade värld behöver svensk sjukvårdspersonal anpassa sig till en ny kontext, där de exponeras för patienter med infektionssjukdomar och ärftliga sjukdomar som tidigare varit ovanliga, försvunnit eller aldrig funnits i vårt sjukdomspanorama. En av dessa sjukdomar är familjär medelhavsfeber, som är viktig att ha i åtanke i länder som tar emot ett stort antal invandrare från östra Medelhavsområdet, t ex Tyskland och Sverige. Sjukdomen är sannolikt vanligare i Sverige än många av oss är medvetna om, och prognosen är god om den behandlas adekvat.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Tre exempel på sjukdomspresentation
Sabina var 8 år och hade attacker med feber och svåra buksmärtor under 1–3 dagar. Attackerna började när hon var 3 år och var förknippade med förhöjt CRP-värde. Sabinas föräldrar var från Libanon. Den genetiska analysen visade på endast en mutation, i stället för två som förväntat vid en autosomalt recessiv sjukdom.
Yusuf var 12 år gammal och lades in på barnavdelning på grund av feber och andningskorrelerade högersidiga bröstsmärtor. Röntgen visade en bild som vid pleurit, och CRP var 115 mg/l. På ronden dagen efter inläggningen var han symtomfri och ville gå hem. Det visade sig att han under de senaste åren haft flera likadana attacker, förknippade med buksmärtor.
Ali var 45 år och sökte på vårdcentral på grund av återkommande buksmärtor. En vanlig urinsticka visade att han hade äggvita i urinen som ett första tecken på njuramyloidos, ett ofta irreversibelt tillstånd.
Fakta 2. Förenklade Tel Hashomer-kriterier [28]
Huvudkriterier1
- Typiska attacker:
– Peritonit (generaliserad)
– Pleurit (unilateral) eller perikardit
– Monoartrit (höft, knä, ankel, fotled)
– Endast feber
- Ofullständiga abdominala attacker
Sekundära kriterier
- Ofullständiga attacker som drabbar en eller båda av följande lokaler:
– Bröstet
– Lederna
- Ansträngningsutlösta smärtor från benen
- Svarar på behandling med kolkicin
1 För diagnos krävs minst ett huvudkriterium eller minst två sekundära kriterier. Typiska attacker definieras som återkommande (minst tre av samma typ), feber (≥38° C) och korta (12 timmar till tre dagar).
Fakta 3. Kriterier för diagnos hos barn [29]
Förekomst av två av fem kriterier och
≥3 attacker talar för familjär medelhavsfeber
- Feber (>38°C, attack med 6–72 timmars duration)
- Magsmärta (peritonit)
- Bröstsmärta (pleurit)
- Artrit
- Ärftlighet för familjär medelhavsfeber
Referenser
- Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140(6):784-90.
- Sohar E, Gafni J, Pras M, et al. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med. 1967;43(2):227-53.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med. 1972;287(25):1302.
- Akar S, Yuksel F, Tunca M, et al. Familial Mediterranean fever: risk factors, causes of death, and prognosis in the colchicine era. Medicine (Baltimore). 2012;91(3):131-6.
- Twig G, Livneh A, Vivante A, et al. Mortality risk factors associated with familial Mediterranean fever among a cohort of 1.25 million adolescents. Ann Rheum Dis. 2014;73(4):704-9.
- Wekell P, Friman V, Balcı-Peynircioğlu B, et al. Familial Mediterranean fever – an increasingly important childhood disease in Sweden. Acta Paediatr. 2013;102(2):193-8.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum. 2009;61(10):1447-53.
- Özen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol. 2014;10(3):135-47.
- Önen F, Sümer H, Türkay S, et al. Increased frequency of familial Mediterranean fever in Central Anatolia, Turkey. Clin Exp Rheumatol. 2004;22(4 Suppl 34):S31-3.
- Masters SL, Simon A, Aksentijevich I, et al. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol. 2009;27:621-68.
- Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. International FMF Consortium. Cell. 1997;90(4):797-807.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet. 1997;17(1):25-31.
- Aksentijevich I, Kastner DL. Genetics of monogenic autoinflammatory diseases: past successes, future challenges. Nat Rev Rheumatol. 2011;7(8):469-78.
- Giancane G, Ter Haar NM, Wulffraat N, et al. Evidence-based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis. 2015;74(4):635-41.
- Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum. 2009;60(6):1862-6.
- Shohat M, Halpern GJ. Familial Mediterranean fever – a review. Genet Med. 2011;13(6):487-98.
- de Jesus AA, Canna SW, Liu Y, et al. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol. 2015;33:823-74.
- Touitou I. The spectrum of familial Mediterranean fever (FMF) mutations. Eur J Hum Genet. 2001;9(7):473-83.
- Jesus AA, Goldbach-Mansky R. IL-1 blockade in autoinflammatory syndromes. Annu Rev Med. 2014;65:223-44.
- Holzinger D, Kessel C, Omenetti A, et al. From bench to bedside and back again: translational research in autoinflammation. Nat Rev Rheumatol. 2015;11(10):573-85.
- Xu H, Yang J, Gao W, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature. 2014;513(7517):237-41.
- Özen S, Demirkaya E, Amaryan G, et al. Paediatric Rheumatology International Trials Organisation; Eurofever Project. Results from a multicentre international registry of familial Mediterranean fever: impact of environment on the expression of a monogenic disease in children. Ann Rheum Dis. 2014;73(4):662-7.
- Kaşifoğlu T, Cansu DU, Korkmaz C. Frequency of abdominal surgery in patients with familial Mediterranean fever. Intern Med. 2009;48(7):523-6.
- Mor A, Gal R, Livneh A. Abdominal and digestive system associations of familial Mediterranean fever. Am J Gastroenterol. 2003;98(12):2594-604.
- Henderson C, Goldbach-Mansky R. Monogenic autoinflammatory diseases: new insights into clinical aspects and pathogenesis. Curr Opin Rheumatol. 2010;22(5):567-78.
- Lachmann HJ, Sengül B, Yavuzsen TU, et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology (Oxford). 2006;45(6):746-50.
- Önen F. Familial Mediterranean fever. Rheumatol Int. 2006;26(6):489-96.
- Livneh A, Langevitz P, Zemer D, et al. Criteria for the diagnosis of familial Mediterranean fever. Arthritis Rheum. 1997;40(10):1879-85.
- Yalçinkaya F, Özen S, Özçakar ZB, et al. A new set of criteria for the diagnosis of familial Mediterranean fever in childhood. Rheumatology (Oxford). 2009;48(4):395-8.
- Nilsson Å. Efterkrigstidens invandring och utvandring. Stockholm: Statistiska centralbyrån; 2004. Demografiska rapporter 2004:5.
- Livneh A, Langevitz P, Zemer D, et al. The changing face of familial Mediterranean fever. Semin Arthritis Rheum. 1996;26(3):612-27.
- Tunca M, Akar S, Önen F, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nationwide multicenter study. Medicine (Baltimore). 2005;84(1):1-11.
Summary
Familial Mediterranean fever – an important disease in a globalised world
Familial Mediterranean fever (FMF) is characterized by recurrent febrile attacks during 1/2–3 days associated with peritonitis, pleuritis and arthritis. FMF is the most common monogenic autoinflammatory disease in the world, with over 100 000 affected individuals. It is particularly common in individuals with an origin in the eastern Mediterranean Basin, where the disease has a prevalence of 100–200 per 100 000. The gene for FMF (MEFV) was identified in 1997 with an autosomal recessive inheritance; however, a significant proportion (≈25%) of clinical patients lack two mutations. MEFV codes for the protein pyrin, whose exact function still needs to be defined. The most serious complication of FMF is amyloid A amyloidosis, in particular renal amyloidosis. FMF is efficiently treated with daily doses of colchicine resulting in an almost normal life expectancy and amyloidosis confined to non-compliant patients. In today’s globalized world we need to adapt to a new context that includes inherited conditions, which have historically been uncommon in our part of the world. One of these conditions is FMF, that should primarily be suspected in individuals with an origin in the eastern Mediterranean Basin and recurrent attacks of fever.


















