De senaste 10 åren har mer än 3 000 nya läkemedel godkänts i Sverige.
Läkemedelsverket är aktivt i det europeiska samarbetet kring godkännande av nya läkemedel. Här ges en kort översikt av godkännandeproceduren.
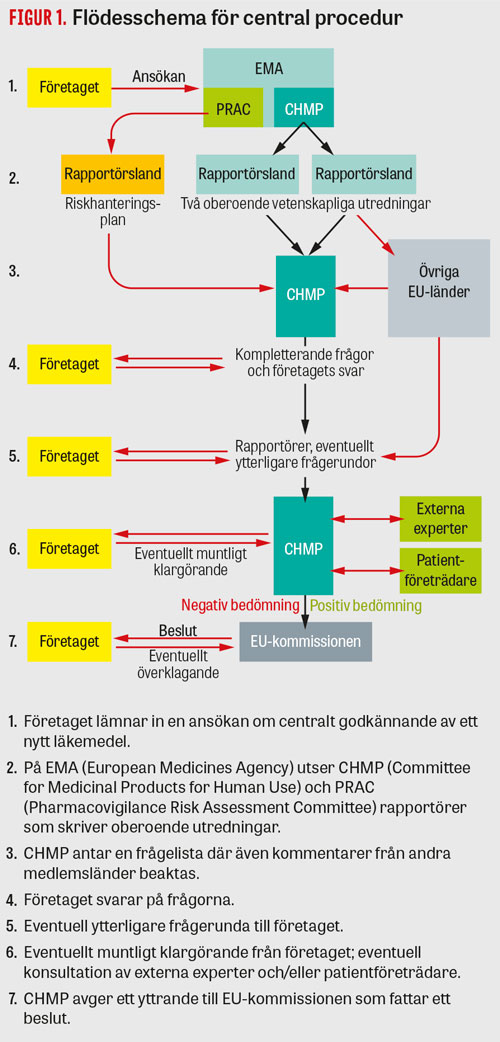
Figur 1
Det regulatoriska systemet för läkemedel inom EU bygger på samverkan mellan nationella myndigheter (såsom Läkemedelsverket) och den europeiska läkemedelsmyndigheten (European Medicines Agency, EMA). De nationella myndigheterna står för den vetenskapliga värderingen av det nya läkemedlet och kommunikationen på nationell nivå. Vetenskapliga kommittéer med nationella delegater har regelbundna möten på EMA som också koordinerar arbetet mellan de nationella myndigheterna. EU-kommissionen ansvarar för det legala regelverket och fattar, efter yttrande från EMA, det formella beslutet om godkännande av läkemedel som utretts via den centrala godkännandeproceduren.
Den avgörande nytta–riskbalansen
Godkännanden av nya läkemedel baseras nästan alltid på gemensamma beslut fattade av flera eller alla EU-länder. De grundläggande kraven för godkännande utgår från ett EU-direktiv som kräver att det sökande företaget visar att nytta–riskbalansen är positiv, det vill säga den sammanvägda bedömningen av nyttan överväger den sammanvägda bedömningen av riskerna för den patientgrupp som definieras av indikationen för läkemedlet. För läkemedel avsedda som en del av en kurativt syftande cancerbehandling kan nyttan överväga riskerna trots förhållandevis grava biverkningar. Däremot tolereras i princip inga allvarliga biverkningar för till exempel ett nytt läkemedel för barn mot hosta.
Underlag för ett godkännande
Godkännandeproceduren är i regel den sista fasen av en 10–15 år lång läkemedelsutvecklingsprocess hos företaget. Ansökan innehåller vetenskaplig dokumentation inom följande tre huvudområden:
- Kemisk/farmaceutisk kvalitet. Data om såväl den aktiva substansens som beredningsformens kvalitet och funktion samt tillverkningsprocesser och kontrollmetoder.
- Preklinisk farmakologi och toxikologi (in vitro-studier, djurmodeller). Den prekliniska dokumentationen är speciellt viktig inom områden som är svåra att studera i människa. Det kan till exempel gälla eventuell fosterskadande effekt eller risk för cancerutveckling.
- Kliniska studier i friska frivilliga och tänkt patientpopulation. Dokumentation av farmakodynamik, farmakokinetik, effekt och biverkningsmönster i människa. Avgörande är i regel de kontrollerade och randomiserade kliniska studier i den tänkta patientpopulationen (fas 3-studier) som syftar till att ge robusta mått på effektstorlek och visa en acceptabel säkerhetsprofil.
Det finns inget generellt krav på att ett nytt läkemedel ska vara bättre än redan godkända alternativ. Därför kan ett nytt läkemedel med mindre uttalad effekt på gruppnivå godkännas om det har en annan säkerhetsprofil än redan godkända alternativ, förutsatt att nytta–riskbalansen för läkemedlet i fråga är positiv.
En riskhanteringsplan fastställs för varje läkemedel vid godkännandet. Den beskriver bland annat hur företaget åläggs att följa upp de identifierade och potentiella riskerna för det nya läkemedlet samt hur dessa bör minimeras vid klinisk användning. I vissa fall är uppföljande studier ett villkor för godkännandet. Kunskapsunderlaget ökar kontinuerligt efter godkännandet och myndigheterna övervakar att läkemedel behåller positiv nytta–riskbalans under hela sin livscykel.
Central procedur – godkännande i hela EU
Då läkemedelsföretaget ansöker om ett godkännande för sitt nya läkemedel i hela EU använder de den centrala godkännandeproceduren (Figur 1). För bioteknologiska produkter, särläkemedel (orphan drugs) samt läkemedel mot cancer, hiv-infektion, neurodegenerativa sjukdomar, diabetes, autoimmuna sjukdomar och virussjukdomar måste den centrala proceduren användas.
Ansökan skickas till EMA, där den vetenskapliga kommittén för humanläkemedel, CHMP (Committee for Medicinal Products for Human Use), fördelar ärendet till två länder som utifrån kompetens och nationella prioriteringar anmält intresse för att utreda och vara så kallat rapportörsland. Även en rapportör från en annan av EMA:s vetenskapliga kommittéer, PRAC (Pharmacovigilance Risk Assessment Committee) utses, som ger stöd i bedömning av riskhanteringsplan och som kommer att ansvara för att följa säkerhet när läkemedlet är på marknaden. Läkemedelsverket anlitas ofta som rapportör för nya läkemedel och ansvarar för ca 20 av totalt ett hundratal rapportörskap per år (Fakta 1) [1].
CHMP består av delegater från varje EU-land samt fem oberoende experter som kompetensmässigt täcker in viktiga ämnesområden. De totalt 33 ordinarie ledamöterna har varsin röst och beslut fattas med enkel majoritet. Norge och Island deltar men saknar rösträtt.
Rapportörsländerna utreder ansökan oberoende av varandra och skriver varsin rapport. Övriga medlemsstater kommenterar rapporterna och vid möten i CHMP enas man om en gemensam rapport med en frågelista gällande oklarheter eller svagheter i företagets dokumentation, vilken företaget ska besvara inom 3–6 månader. Om företaget besvarar frågorna adekvat och nytta–riskbalansen bedöms vara positiv avger CHMP ett positivt yttrande. Om oklarheter alltjämt kvarstår startas en ny frågerunda med kompletterande frågor; detta kan göras två gånger. Indikationen föreslås initialt av företaget men diskuteras under utredningens gång och kan ändras till att bli både snävare eller bredare beroende på i vilken population nytta–riskbalansen bedöms vara positiv. Om CHMP slutligen bedömer att det inte går att hitta en population för vilken nytta–riskbalansen är positiv och avslår ansökan kan företaget begära en omvärdering av underlaget, och då utses två andra rapportörsländer som genomför en förnyad bedömning. Under hela utvärderingsprocessen kan CHMP rådfråga externa expertgrupper, dock efter en strikt bedömning av eventuella jäv. Det blir också allt vanligare att delar av ansökan diskuteras med patientföreträdare. Normalt tar en utredning ungefär 18 månader (varav myndigheterna har 210 dagars utredningstid) från ansökan till beslut om godkännande av EU-kommissionen, vilket gäller i hela EU. De flesta ansökningar leder till ett godkännande och endast ett mycket litet antal får avslag. Förra året, 2018, beslutade CHMP om 84 positiva rekommendationer för godkännande och 5 negativa rekommendationer; 10 ansökningar drogs tillbaka av företagen [2].
För läkemedel som är avsedda för livshotande eller svårt handikappande tillstånd, akutläkemedel eller läkemedel mot mycket sällsynta sjukdomar har det sedan 2006 varit möjligt att söka ett villkorat godkännande. För ett sådant måste nytta–riskbalansen bedömas vara positiv på de begränsade data som finns vid tidpunkten för godkännandet, och det måste vara troligt att konklusiva data kan komma att presenteras. Konfirmerande studieresultat utgör villkoret för det tidiga godkännandet. Läkemedlet ska också svara mot ett ej uppfyllt medicinskt behov och riskerna med att inte göra läkemedlet tillgängligt ska väga tyngre än riskerna som finns med att godkänna på begränsade data. Under de första 10 åren godkände EMA 30 sådana läkemedel (1–4 per år) varav 11 läkemedel har uppfyllt sina villkor, 2 dragits tillbaka av företagen och 17 fortfarande har pågående villkorade studier [3].
Nationell, ömsesidig och decentraliserad procedur
Det kan finnas olika skäl till att ett läkemedelsföretag väljer att inte ansöka om ett centralt godkännande, till exempel att företaget bara finns representerat i vissa EU-länder. I andra fall kan det vara medicinska eller sociala skäl, som den varierande synen på »dagen efter-piller« i olika länder.
I den nationella proceduren ansöker företaget om godkännande i ett EU-land, vars läkemedelsmyndighet då gör hela utredningen.
I den ömsesidiga proceduren hänvisar företaget till ett land där läkemedlet redan godkänts (referensland) och väljer sedan i vilka ytterligare EU-länder som ansökan görs. Respektive nationell myndighet tar sedan ställning till referenslandets tidigare utredning.
Den decentraliserade proceduren är en variant på den ömsesidiga proceduren. Skillnaden är att denna ansökan från början går parallellt till de inblandade länderna och ett utvalt land agerar referensland.
På apotekshyllan – tiden efter godkännandet
I samband med att ett nytt läkemedel godkänns sammanställs en produktinformation som ger vägledning om bland annat terapeutisk indikation, kontraindikationer, dosering, användning under graviditet och amning, varningar, biverkningar samt en sammanfattning av de viktigaste studieresultaten. För centralt godkända läkemedel sammanställs även en mer omfattande sammanfattning av de data som legat till grund för ansökan samt CHMP:s bedömning av underlaget i en så kallad EPAR (European public assessment report). EPAR innehåller en sammanställning av företagets egna data som kan vara mer omfattande än de studier som finns publicerade för det nya läkemedlet. Båda dessa dokument publiceras på EMA:s webbplats och är tillgängliga för allmänheten [4]. Via Läkemedelsverkets webbplats hittas produktinformationen på svenska för alla i Sverige godkända läkemedel, och motsvarande information återges även i Fass [5, 6]. Läkemedelsverket publicerar ofta en monografi på webbplatsen, samt i verkets tidskrift, med en vetenskaplig värdering av det nya läkemedlet i samband med att det godkänns.
Hur det nya läkemedlet faktiskt används i varje enskilt land påverkas även av nationella behandlingsrekommendationer och förmånssystem. Kostnadseffektiviteten vägs inte in vid godkännandet. I Sverige är det Tandvårds- och läkemedelsförmånsverket (TLV) som gör en hälsoekonomisk värdering i beaktande av riksdagens tre etiska principer (människovärdes-, solidaritets-/behovs- och kostnadseffektivitetsprincipen) och bestämmer om läkemedlet ska vara rabatterat och omfattas av högkostnadsskyddet. TLV förser också NT-rådet (nya terapier) vid Sveriges Kommuner och landsting med hälsoekonomiska underlag gällande nationella rekommendationer för läkemedel som används på sjukhus. Läkemedelsverket kan besluta att läkemedel ska vara utbytbara om de har bedömts som medicinskt likvärdiga vad gäller sammanvägd effekt och säkerhet [7].
För vissa läkemedel kan företagen ansöka om receptfrihet (OTC, försäljning över disk), vilken beslutas nationellt, i Sverige av Läkemedelsverket. Receptfria läkemedel är avsedda för egenvård, därför måste indikationen och säkerhetsprofilen vara lämplig för självmedicinering. Vanligen krävs några års användning med receptbeläggning innan receptfrihet kan diskuteras.
Läkemedelsverkets arbetssätt
Tillståndsgivningen på Läkemedelsverket sysselsätter drygt 300 personer. Flera olika yrkesgrupper i team utreder gemensamt en ansökan om godkännande, och många av utredarna är disputerade inom olika områden. Av de kliniska utredarna är ett 70-tal läkare, de flesta med bakgrund inom läkemedelsintensiva specialiteter. Alla utredningar gällande nya läkemedel kvalitetsgranskas i flera steg internt på Läkemedelsverket, först inom varje område för sig (farmaceutisk kvalitet, preklinik, farmakokinetik samt klinik), därefter i en gemensam multidisciplinär kvalitetsgranskning av seniora representanter från varje område. Detta arbetssätt används även i andra ärenden och utnyttjar Läkemedelsverkets samlade kompetens, vilket bidrar till en hög och jämn nivå på utredningsrapporterna.
Läkemedelsverket har ett gott anseende i det europeiska samarbetet. Vårt engagemang på europeisk nivå möjliggör viktig kunskapsöverföring till och från Sverige om läkemedel och läkemedelsanvändning, vilket gagnar svenska patienter.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Läkemedelsgodkännanden i Sverige [1, 2]
Totalt finns ca 13 000 läkemedel för humant bruk godkända i Sverige. Antalet unika substanser är ca 2 000 och i den centrala proceduren tillkom 42 nya aktiva substanser under 2018.
2017 godkändes 286 läkemedelsprodukter.
2017 var Sverige rapportör eller referensland för:
- 17 läkemedel i den centrala proceduren (rapportörskap)
- 106 läkemedel i den ömsesidiga/decentraliserade proceduren (referensland)
- 46 läkemedel i den nationella proceduren.
Referenser
- Läkemedelsverket. Årsredovisning 2017.www.lakemedelsverket.se/upload/om-lakemedelsverket/publikationer/Arsredovisning-och-miljoredovisning/Lakemedelsverkets-arsredovisning-2017.pdf
- Human medicines highlights 2018. London: European Medicines Agency (EMA); 2018.
- European Medicines Agency (EMA); Sebris Z. Conditional marketing authorisation. Report on ten years of experience at the EMA. 27 jun 2017. www.ema.europa.eu/documents/presentation/presentation-conditional-marketing-authorisation-report-ten-years-experience-ema-zigmars-sebris-ema_en.pdf
- European Medicines Agency (EMA). European public assessment reports (EPAR). https://www.ema.europa.eu/en/medicines/download-medicine-data#european-public-assessment-reports-(epar)-section
- Läkemedelsverket. Läkemedelsfakta. www.lakemedelsverket.se/LMF/
- Läkemedelsindustriföreningen. Farmaceutiska specialiteter i Sverige (Fass). www.fass.se/LIF/startpage
- Läkemedelsverket. Kriterier för utbytbarhet. 1 jul 2016 [citerat 1 feb 2019]. www.lakemedelsverket.se/malgrupp/Halso—sjukvard/Forskrivning/Utbytbara-lakemedel-/Kriterier-for-utbytbarhet/