



Ataxier utgör en heterogen grupp sjukdomar med överlappande symtom.
Vid utredningen är det viktigt att söka behandlingsbara tillstånd såsom neurometabola och autoimmuna sjukdomar.
Snabb progress kännetecknar ofta paraneoplastisk cerebellär degeneration, autoimmun sjukdom eller multipel systematrofi.
Tillämpning av ny DNA-sekvensteknologi möjliggör preciserad diagnostik för allt fler patienter, vilket kan vara avgörande för prognos och behandling.
Vid ärftliga former av ataxi är säkerställda genetiska orsaker en förutsättning för genetisk vägledning.
Ataxi kännetecknas av bristande balans och koordination orsakad av påverkan på lillhjärnans motoriska funktioner. Vestibulär påverkan och polyneuropatier kan också medföra ataxi i varierande omfattning, men den här översikten omfattar cerebellära sjukdomar. Antalet sjukdomar med ataxi som del- eller huvudsymtom är över hundra, vilket illustrerar den diagnostiska utmaningen för neurologer [1]. Ataxi kommer från grekiskans negation »a-taxis« och betyder »avsaknad av ordning«. De karakteristiska symtomen utgörs av störd finmotorik, ostadighet, sluddrigt tal, dysfagi och/eller störda sackader.
Lillhjärnan är en evolutionärt konserverad del av nervsystemet och finns hos alla ryggradsdjur. Lillhjärnan har projektioner till alla strukturer av betydelse för motorisk funktion [2, 3]. Den tar emot afferent information från flera delar av cerebrala kortex (sensorimotoriska arean, frontala, parietala och temporala regioner), ryggmärgen (spinocerebellära banor), vestibulära nerven och hjärnstammen [2, 3]. Lillhjärnan tar även emot information från delar av det av limbiska systemet och autonoma centrum (hypotalamus), modulerar precisionen i rörelserna och är således viktig för balans, kroppshållning och koordination. Lillhjärnan spelar också en viktig roll vid motorisk planering och inlärning, kontroll av ögonmotoriken och tal. Utifrån funktion består lillhjärnan av tre delar: vermis, intermediär zon (paravermis) och lateral zon [2]. Påverkan på den vermala delen ger huvudsakligen symtom i form av axial ataxi, okulomotoriska avvikelser, dysartri och titubation (skakningar). Påverkan på laterala delarna är däremot kopplad till framträdande dysmetri (nedsatt koordination), tremor (aktions- och kinetisk) och nedsatt diadokokinesi. Paravermal påverkan är huvudsakligen associerad med dysartri.
Lillhjärnan har även betydelse för olika kognitiva funktioner, beteende och emotioner, vilket förklarar de betydande icke-motoriska symtom som kan drabba patienter med ataxi [2-5]. Vid komplexa ataxier kan även extracerebellära motorikstörningar uppträda i form av parkinsonism och dystoni, liksom andra neurologiska symtom som ögonmotorikstörning (långsamma ögonrörelser eller oftalmoplegi), synpåverkan (katarakt, retinala förändringar, mm), dysautonomi (urinträngningar, inkontinens eller ortostatism), epilepsi, spasticitet och/eller polyneuropati i varierande omfattning.
Vid systemsjukdomar kan endokrina rubbningar, cancer eller kardiella manifestationer uppträda tillsammans med ataxi. Vid hereditära och neurodegenerativa ataxisjukdomar är förloppet progressivt med gradvis förlust av funktioner och olika förmågor. Ett fåtal ataxisjukdomar är behandlingsbara, och patienter med ataxi av oklar etiologi bör alltid utredas för att precisera orsaken och för önskad effekt av eventuell behandling [1].
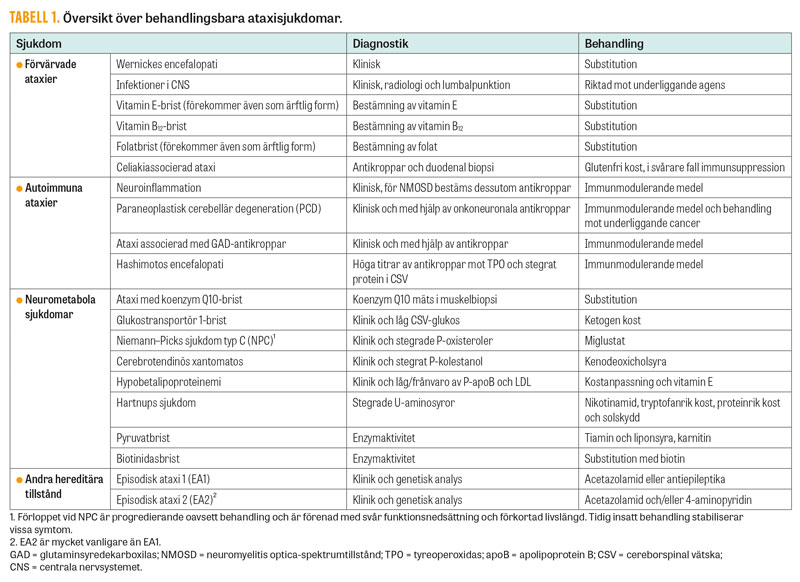
Ett exempel på detta är vitamin E-brist, som förekommer både i förvärvad och ärftlig form. Båda formerna kan behandlas med tokoferylacetat, och utfallet beror på när i sjukdomsförloppet behandlingen inleds. Farmakologisk behandling vid ataxi saknas dock för den absoluta majoriteten av patienter, och undantagen är få (Tabell 1). Däremot ska det poängteras att manifestationer vid vissa former av ataxi, såsom spasticitet, urinträngningar och parkinsonism, kan behandlas farmakologiskt.
Förenklat kan man dela upp ataxisjukdomar i tre huvudgrupper:
- förvärvade (icke-ärftligt betingade)
- neurodegenerativa
- familjära (ärftligt betingade) tillstånd [6].
Utifrån den dominerande fenomenologin betecknas en ataxi som axial om gången är mer påverkad än koordinationen, och appendikulär om finmotoriken är mer drabbad än balansen.
Prevalens
Prevalensen av ataxi hos barn, huvudsakligen ärftliga former, uppskattas till cirka 26/100 000 i europeiska populationer [7]. Den totala prevalensen av ärftligt betingad cerebellär ataxi uppskattas till 0,5–5,6/100 000 personer, med avsevärda variationer i olika världsdelar [8]. Den hittills största kartläggningen av etiologiska faktorer vid ataxi är baserad på en kohort som inkluderade 1 500 patienter i Sheffield, England. I studien utgjorde glutenataxi 25 procent av fallen, ärftlig ataxi 13 procent, alkoholcerebellopati 12 procent, cerebellär multipel systematrofi (MSA-C) 11 procent och paraneoplastisk cerebellär degeneration (PCD) 3 procent. Etiologin hos upp till 24 procent av patienterna bedömdes vara idiopatisk [9]. Det är värt att notera att centrumet i Sheffield har varit mycket tongivande i etableringen av glutenataxi som entitet.
Förvärvade ataxier
Debutsymtom, ålder och progressionstakt är vägledande vid utredning. Akut debut talar för cerebrovaskulär skada, neuroinflammation, intoxikation (vissa antiepileptika, litium, droger och tungmetaller), infektion i centrala nervsystemet (CNS) eller parainfektiös cerebellit. Svår tiaminbrist (vitamin B1) hos individer med alkoholmissbruk eller andra riskfaktorer (exempelvis kakexi, anorexi och malabsorption) leder till akut ataxi som ett led i Wernickes encefalopati. Andra symtom vid akut tiaminbrist inkluderar konfusion och varierande grad av ögonmuskelpareser. Omedelbar substitution reverserar bilden. Upprepade episoder med obehandlad Wernickes encefalopati leder till det irreversibla Korsakoffs syndrom. Subakut debut är kännetecknande för paraneoplastisk cerebellär degeneration, tumörer, superficiell sideros och prionsjukdom.
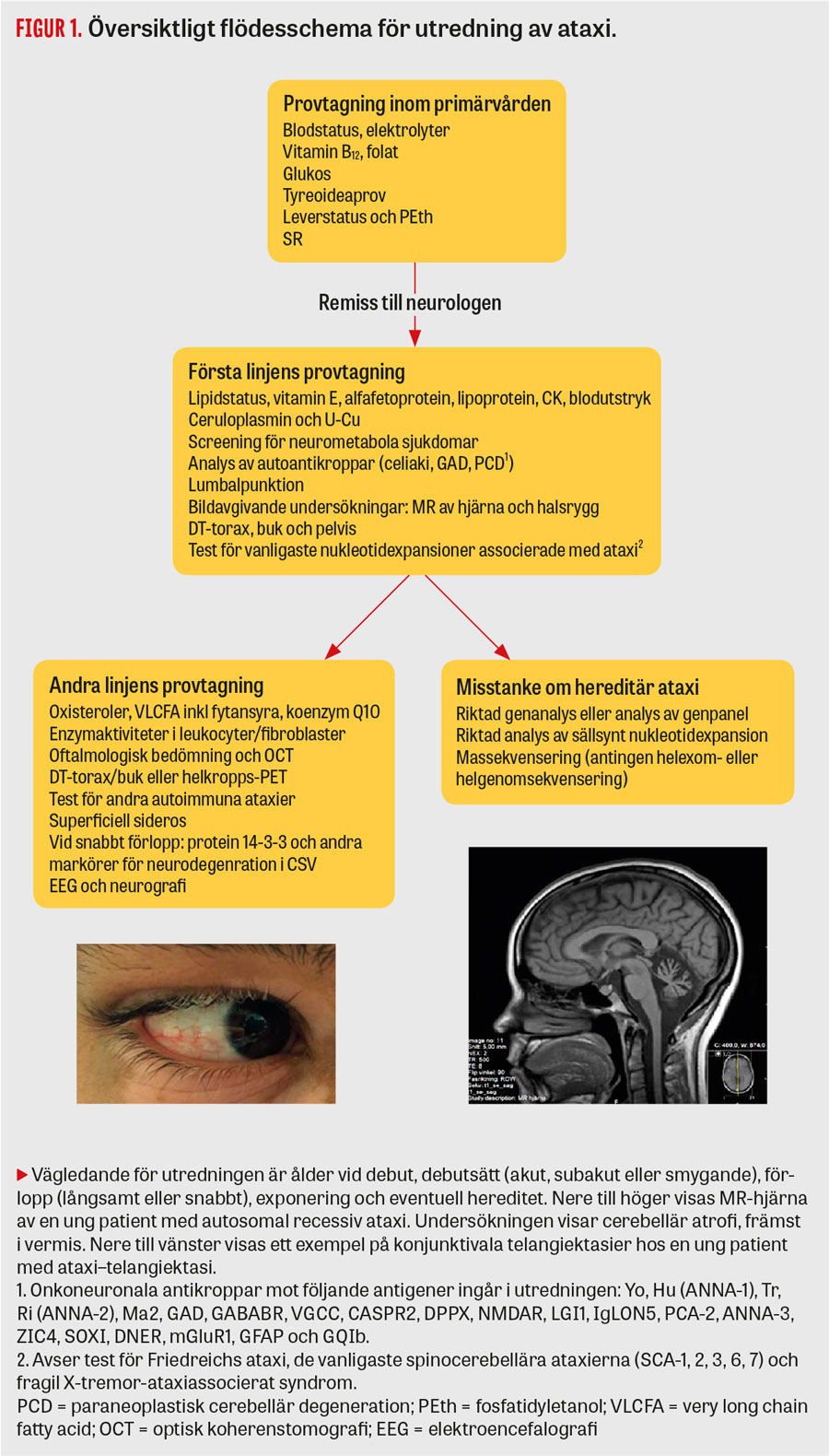
Förloppet vid paraneoplastisk cerebellär degeneration är oftast snabbt och leder till höggradig funktionsnedsättning inom veckor–månader. Avgörande för överlevnaden är att påvisa och behandla underliggande malignitet, och behandlingsförsök med steroider rekommenderas tidigt i förloppet. Vid terapeutiskt svar kan annan immunmodulerande behandling prövas. Paraneoplastisk cerebellär degeneration förekommer vid flera cancerformer men är oftast associerad med lung-, bröst- och ovarialcancer samt Hodgkins lymfom [6]. Vid misstanke bör patienten screenas med onkoneuronala antikroppar (Figur 1) [10], riktad radiologi och lumbalpunktion. Det bör noteras att antikropparna inte är specifika för någon cancerform.
Smygande debut av ataxi kan ha många tänkbara etiologier och en systematisk kartläggning fordras. Alkoholmissbruk, kronisk exponering för toxiska substanser (läkemedel och vissa droger), celiaki och neurodegenerativa sjukdomar är exempel på etiologi till smygande kroniska ataxier (se nedan) [1, 6, 11, 12]. En smygande debut är också karakteristisk för majoriteten av hereditära ataxier.
Alkoholutlöst cerebellopati är vanligt förekommande hos individer med kroniskt alkoholberoende. Ataxin är då axial, och neuroradiologin visar i det typiska fallet vermisatrofi. I dessa fall bidrar ofta näringsbrist, främst tiaminbrist, till den alkoholrelaterade ataxin [6]. Vitamin B12-brist ska heller inte missas. Tillståndet kännetecknas av sensorisk ataxi (det vill säga afferenta banor till cerebellum är påverkade), pyramidala symtom och ibland kognitiva och psykiska symtom. Obehandlad är vitamin B12-brist förenad med bestående bortfallssymtom. Det är även viktigt att bestämma vitamin E-nivåer hos individer med malabsorption och/eller kakexi. Flera studier har visat ett samband mellan celiaki och ataxi, och strikt glutenfri kost är då ofta effektiv [9, 13].
Snabb progressionstakt är alltid ett varningstecken oavsett ålder. Typfallet är autoimmuna tillstånd i bred bemärkelse, paraneoplastisk cerebellär degeneration, ataxi associerad med antikroppar mot proteinet GAD (glutaminsyradekarboxylas), infektioner i CNS och parainfektiösa processer, men snabb progress förekommer även vid neurodegenerativa sjukdomar.
Neurodegenerativa ataxier
Framträdande ataxi hos individer över 30 år som utvecklar dysautonomi och/eller parkinsonism kännetecknar den cerebellära formen av multipel systematrofi, MSA-C. När parkinsonism dominerar symtombilden betecknas tillståndet i stället MSA-P. Multipel systematrofi är, liksom Parkinsons sjukdom, en synukleinopati men svarar tämligen begränsat, om ens alls, på levodopa. En annan viktig skillnad är att multipel systematrofi har en dålig prognos med väsentligt förkortad livslängd [14]. I extremt sällsynta fall kan snabbt progredierande ataxi, kognitiv svikt och myoklonus hos äldre tyda på sporadisk Creutzfeldt–Jakobs sjukdom. Diagnostiken vid neurodegenerativa ataxier är uteslutande klinisk och bygger på en kombination av diagnoskriterier. Vissa bildavgivande modaliteter (MR-hjärna och/eller FDG [fluorodeoxiglukos]-PET) och biokemiska analyser i likvor kan stärka misstanken. Definitiv diagnos av neurodenerativa sjukdomar fordrar neuropatologi och illustrerar behovet av ytterligare biomarkörer inom fältet. Det är viktigt att påpeka att upp till 30 procent av vuxna individer med idiopatisk ataxi utvecklar multipel systematrofi, varför regelbunden uppföljning är viktig.
En egen heterogen subgrupp utgörs av så kallad ILOCA (idiopathic late-onset cerebellar ataxia) som i litteraturen gått under många namn (t ex sporadic adult-onset ataxia of unknown etiology [SAOA]). Gruppen skiljer sig från multipel systematrofi då progressionstakten är långsam och dysautonomi saknas. Sannolikt finns det i den gruppen en andel odiagnostiserade fall med förvärvad eller hereditär ataxi [1, 6, 11, 12].
Hereditära ataxisjukdomar
Samtliga nedärvningsmönster – autosomalt dominant eller recessiv, X-kromosombunden, och mitokondriell – förekommer vid familjär ataxi. Den snabba utvecklingen av ny DNA-sekvensteknologi och dess tillämpning har medfört att ett stort antal gener har associerats med ataxi. Metoden är tillgänglig inom sjukvården och har avsevärt underlättat diagnostiken. Identifiering av genvarianter som orsakar ataxi har gett omfattande och mer ingående förståelse av bakomliggande sjukdomsmekanismer. Genetiken vid olika former av genetiskt betingade ataxier har också avslöjat mekanistiska överlappningar och samband på molekylär nivå. Det har påverkat förlegade kliniska indelningar av sjukdomsgrupper som kan vara svåra att överblicka. Förslag till användarvänlig och modernare nomenklatur har lanserats men är ännu inte fullt etablerad (15-17).
Vid hereditär ataxi är debuten oftast smygande, med undantag för vissa neurometabola sjukdomar, episodiska ataxier och sjukdomar associerade med mutationer i ATP1A3-genen. Neurometabola sjukdomar är en kliniskt och biokemiskt heterogen grupp som debuterar i barndomen och oftast följer ett recessivt nedärvningsmönster [18]. Vid Wilsons sjukdom kan ataxi vara ett delsymtom vid sidan om engagemang av basala ganglier och neuropsykiatriska symtom, men framträdande ataxi är däremot ovanlig.
Patologiska trinukleotidexpansioner i specifika genregioner intar en särskild plats inom neurologin. Samtliga kända expansioner i DNA är nämligen associerade med neurodegeneration eller neuromuskulära sjukdomar. Expansioner av trinukleotiden CAG, som kodar för aminosyran glutamin, är orsak till den vanligaste subgruppen av ärftliga ataxisjukdomar och även till Huntingtons sjukdom (se separat artikel i detta temanummer). Dominant nedärvd spinocerebellär ataxi 3/Machado–Josephs sjukdom (SCA3/MJS), som betingas av en patologisk CAG-expansion i ataxin 3, utmärker sig i gruppen av både epidemiologiska och kliniska skäl [19, 20]. SCA3/MJS debuterar oftast i vuxen ålder och är den vanligaste formen av ärftlig ataxi både globalt [20] och i Sverige [21].
Fragil X-tremor-ataxiassocierat syndrom (FXTAS) är en X-kromosombunden sjukdom som oftast drabbar äldre manliga bärare av en så kallad premutation i FMR1-genen [22]. Premutationen utgörs av en intronisk CGG-expansion, medan en full mutation i FMR1 orsakar fragil X-syndromet.
Friedreichs ataxi är en autosomalt recessiv systemsjukdom som debuterar med kordinationsstörningar i vanligen barnaåren och som progredierar med systemiska manifestationer (kardiomyopati, skolios, risk för diabetes, syn- och hörselnedsättning) [23, 24]. Polyneuropati ingår i syndromet som utgör ett exempel på sensorisk ataxi. Friedreichs ataxi orsakas av en intronisk GAA-expansion i homozygot form och är, till skillnad från i övriga Europa, sällsynt i Skandinavien [25]. Samtliga ataxier orsakade av trinukleotidexpansioner har en progressiv karaktär och är obotliga. En intressant upptäckt inom gruppen ataxier som orsakas av DNA-expansioner vid sent debuterande ataxi med neuropati och vestibulopati (CANVAS) tillkännagavs nyligen [26]. Arbetet visar hur förfinad DNA-metodik resulterat i upptäckt av en patologisk homozygot nukleotidexpansion i RFC1-genen.
Utredning
Utredningen av ataxier bygger på en noggrann genomgång av debutålder, förlopp, ärftlighet, exponeringar (alkoholmissbruk, vissa antiepileptika och behandling med cytostatika) och undersökningsfynd. Patienten undersöks först med en riktad skala för ataxi. Två etablerade skalor finns: Scale for the assessment and rating of ataxia (SARA) samt International cooperative ataxia rating scale (ICARS) [27, 28]. Artikelförfattarna förespråkar SARA eftersom protokollet är enkelt att följa och bara tar några minuter i anspråk. För neurologiska symtom utöver ataxi tillämpas Inventory for non-ataxia symptoms (INAS). INAS är en genomgång av eventuell förekomst av andra cerebellära avvikelser (nystagmus och störda sackader), pyramidala och extrapyramidala fenomen samt ögonmotorikpåverkan (långsamma sackader eller oftalmoplegi). Förekomst av kutana stigmata kan vara vägledande, till exempel telangiektasier i konjunktiva vid ataxi–telangiektasi.
Efter klinisk undersökning och screening med basala prov inom primärvården ska patienten remitteras till en neurologklinik för fortsatt utredning och behandling (Tabell 1). Ytterligare utredning omfattar då provtagning för biokemiska analyser, olika bildavgivande modaliteter och neurofysiologiska test. Oavsett ålder och debutform ska patienter med ataxi genomgå kartläggning av hjärna och halsrygg med bildavgivande metod, helst MR. Värt att notera är att avsaknad av familjehistoria inte utesluter ärftlig ataxi. Autosomalt recessiva sjukdomar, nymutationer i gener med dominant effekt, nedsatt penetrans och adoption kan vara förklaringen till avsaknad av hereditet.
Utredningen vid neurologklinik innefattar stegvisa provtagningar och i förekommande fall lumbalpunktion och kartläggning med neurofysiologiska test. Provtagningen inkluderar screening för neurometabola sjukdomar (exempelvis enzymaktivitet) och, vid misstanke, autoantikroppar för celiaki, paraneoplastisk cerebellär degeneration, GAD, andra autoantikroppar och eventuell muskelbiopsi. Onkoneuronala antikroppar mot följande antigener ingår i utredningen: Yo, Hu (ANNA-1), Tr, Ri (ANNA-2), Ma2, GAD, GABABR, VGCC, CASPR2, DPPX, NMDAR, LGI1, IgLON5, PCA-2, ANNA-3, ZIC4, SOXI, DNER, mGluR1, GFAP och GQ1b [10]. Andra mycket ovanliga antikroppar kan ibland bli aktuella [10]. I cerebrospinal vätska analyseras vanligen följande rutinparametrar: celler, protein, cytologi, fraktionerade proteiner (oligoklonala band), laktat och glukos [1].
Utifrån förlopp och misstanke analyseras för autoantikroppar, protein 14-3-3 och/eller markörer för neurodegeneration (totalt tau, fosforylerat tau och beta-amyloid) och nervskademarkören neurofilament light (NFL) protein. Polyneuropati hos personer med ataxi stärker misstanken om ärftlig sjukdom och indikationen för provtagning [1, 12].
Vid sporadisk ataxi blir genetiska analyser aktuella först när förvärvade former av ataxi uteslutits (Figur 1). Vanligen börjar man med gentest riktade mot de vanligaste patologiska nukleotidexpansionerna. Om patienten förblir odiagnostiserad blir nästa steg antingen helexomsekvensering eller helgenomsekvensering.
Neuroradiologi, allra helst MR av hjärna och halsrygg, kan bidra med diagnostiska ledtrådar och utesluta strukturella skador (som till exempel stroke, tumörer, neuroinflammation eller superficiell sideros). Observera att det inte finns någon korrelation mellan grad av cerebellär ataxi och atrofi. Signalförändringar i hjärnstammen och i cerebellära pedunklar kan peka på en viss diagnos men är inte specifika. Exempelvis är så kallat »hot cross bun«-tecken (korsformigt utseende i mellersta hjärnstammen), som ses hos en del patienter med multipel systematrofi, men avvikelsen är inte specifik.
Ny DNA-sekvensteknologi har underlättat utredningarna vid misstanke om ärftlig ataxi. Metoden har blivit alltmer tillgänglig, men utmaningar ligger i tolkningen av nya genetiska varianter (varianter av oklar betydelse) och behovet av nya tekniker för att detektera patologiska nukleotidexpansioner. En lovande metod för att identifiera DNA-expansioner har nyligen etablerats i form av så kallad »long-read sequencing«, som prövats i tertiära centrum.
Olika utredningsalgoritmer har föreslagits som anpassats beroende på tillgänglighet till massekvensering [29-31]. I samtliga riktlinjer betonas vikten av att fånga behandlingsbara tillstånd i utredningen [1,12, 18].
Behandling
Omhändertagande av patienter med ataxi sker lämpligen i en multidisciplinär grupp – progredierande ataxisjukdomar fordrar regelbunden uppföljning. Ett fåtal ataxisjukdomar är som nämnt behandlingsbara [1, 12, 18] (Tabell 1).
Nykterhet, och i förekommande fall substitution med peroralt vitamin B-komplex, bromsar progressen vid alkoholutlöst cerebellopati. Vid Wernickes encefalopati ges tiamin, medan glutenfri kost ofta är effektiv mot celiakiassocierad ataxi, och i behandlingsrefraktära fall kan immunsuppression bli aktuell [32]. Vid paraneoplastisk cerebellär degeneration är prognosen generellt sett mycket dålig, och avgörande för överlevnaden är att identifiera och behandla den underliggande cancersjukdomen [33]. Immunmodulerande behandling kan stabilisera motorikstörningen, men underlaget för en entydig rekommendation är begränsat [33].
Under 1990-talet tydde vissa studier på att serotonerga substanser kunde vara ett behandlingsalternativ vid ataxi, men studiepopulationerna var små och heterogena [34, 35]. Resultaten kunde inte konfirmeras, och någon farmakologisk behandling riktad mot de motoriska störningarna vid ataxi finns ännu inte [36]. Symtomatisk behandling av andra associerade fenomen kan däremot förbättra livskvaliteten för patienten. Evidens finns för att intensiv fysioterapi under korta perioder är gynnsam. En annan metod med potential är transkraniell magnetstimulering, som uppmärksammats på senare år [36]. Vid sporadisk Creutzfeldt–Jakobs sjukdom är prognosen mycket dyster, och palliativa insatser blir ofta aktuella tidigt i förloppet.
Framtiden
Pågående studier med antisens-oligonukleotider mot Huntingtons sjukdom och som sjukdomsmodifierande substanser vid spinal muskelatrofi och Duchennes muskeldystrofi inger förhoppningar om att skräddarsydda behandlingar kommer att bli verklighet även för hereditära ataxier. Preklinisk forskning med antisens-oligonukleotider pågår för de vanligaste ärftliga ataxierna [37]. Behovet av terapeutiska studier avseende ataxier är stort.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
(uppdaterad 2020-03-16)
Referenser
- de Silva R, Greenfield J, Cook A, et al. Guidelines on the diagnosis and management of the progressive ataxias. Orphanet J Rare Dis. 2019;14(1):51.
- Manto M. Cerebellar disorders. A practical approach to diagnosis and management. Cambridge: Cambridge University Press; 2010. p. 2-51.
- Guell X, Gabrieli JDE, Schmahmann JD. Triple representation of language, working memory, social and emotion processing in the cerebellum: convergent evidence from task and seed-based resting-state fMRI analyses in a single large cohort. Neuroimage. 2018;172:437-49.
- Schmahmann JD, Sherman JC. The cerebellar cognitive affective syndrome. Brain. 1998;121(Pt 4):561-79.
- Ahmadian N, van Baarsen K, van Zandvoort M, et al. The cerebellar cognitive affective syndrome – a meta-analysis. Cerebellum. 2019;18(5):941-50.
- Klockgether T. Sporadic ataxia with adult onset: classification and diagnostic criteria. Lancet Neurol. 2010;9(1):94-104.
- Musselman KE, Stoyanov CT, Marasigan R, et al. Prevalence of ataxia in children: a systematic review. Neurology. 2014;82(1):80-9.
- Ruano L, Melo C, Silva MC, et al. The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology. 2014;42(3):174-83.
- Hadjivassiliou M, Martindale J, Shanmugarajah P, et al. Causes of progressive cerebellar ataxia: prospective evaluation of 1500 patients. J Neurol Neurosurg Psychiatry. 2017;88(4):301-9.
- Balint B, Vincent A, Meinck HM, et al. Movement disorders with neuronal antibodies: syndromic approach, genetic parallels and pathophysiology. Brain. 2018;141(1):13-36.
- Kuo SH. Ataxia. Continuum (Minneap Minn). 2019;25(4):1036-54.
- de Silva RN, Vallortigara J, Greenfield J, et al. Diagnosis and management of progressive ataxia in adults. Pract Neurol. 2019;19(3):196-207.
- Hadjivassiliou M, Grünewald RA, Chattopadhyay AK, et al. Clinical, radiological, neurophysiological, and neuropathological characteristics of gluten ataxia. Lancet. 1998;352(9140):1582-5.
- Figueroa JJ, Singer W, Parsaik A, et al. Multiple system atrophy: prognostic indicators of survival. Mov Disord. 2014;29(9):1151-7.
- Marras C, Lang A, van de Warrenburg BP, et al. Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society task force. Mov Disord. 2017;32(5):724-5.
- Rossi M, Anheim M, Durr A, et al; International Parkinson and Movement Disorder Society Task Force on Classification and Nomenclature of Genetic Movement Disorders. The genetic nomenclature of recessive cerebellar ataxias. Mov Disord. 2018;33(7):1056-76.
- Beaudin M, Matilla-Dueñas A, Soong BW, et al. The classification of autosomal recessive cerebellar ataxias: a consensus statement from the Society for Research on the Cerebellum and Ataxias Task Force. Cerebellum. 2019;18(6):1098-125.
- Ebrahimi-Fakhari D, Van Karnebeek C, Münchau A. Movement disorders in treatable inborn errors of metabolism. Mov Disord. 2019;34(5):598-613.
- GeneReviews; Bird TD. Hereditary ataxia overview. 28 okt 1998 [uppdaterat 25 jul 2019]. https://www.ncbi.nlm.nih.gov/books/NBK1138/
- Klockgether T, Mariotti C, Paulson HL. Spinocerebellar ataxia. Nat Rev Dis Primers. 2019;5(1):24.
- Socialstyrelsen. Ovanliga diagnoser. Spinocerebellära ataxier, dominant ärftliga. 5 maj 2015 [citerat 20 aug 2019]. https://www.socialstyrelsen.se/stod-i-arbetet/ovanliga-diagnoser/spinocerebellara-ataxier-dominant-arftliga/
- GeneReviews; Saul RA, Tarleton JC. FMR1-related disorders. 16 jun 1998 [uppdaterat 26 apr 2012]. https://www.ncbi.nlm.nih.gov/books/NBK1384/
- GeneReviews, Bidichandani SI, Delatycki MB. Friedreich ataxia. 18 dec 1998 [uppdaterat 1 jun 2017]. https://www.ncbi.nlm.nih.gov/books/NBK1281/
- Socialstyrelsen. Ovanliga diagnoser. Friedreichs ataxi. 8 jun 2016 [citerat 20 aug 2019]. https://www.socialstyrelsen.se/stod-i-arbetet/ovanliga-diagnoser/friedreichs-ataxi/
- Wedding IM, Kroken M, Henriksen SP, et al. Friedreich ataxia in Norway – an epidemiological, molecular and clinical study. Orphanet J Rare Dis. 2015;10:108.
- Trouillas P, Takayanagi T, Hallett M, et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J Neurol Sci. 1997;145(2):205-11.
- Schmitz-Hübsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717-20.
- Cortese A, Simone R, Sullivan R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51(4):649-58.
- van de Warrenburg BP, van Gaalen J, Boesch S, et al. EFNS/ENS Consensus on the diagnosis and management of chronic ataxias in adulthood. Eur J Neurol. 2014;21(4):552-62.
- Fogel BL, Lee H, Deignan JL, et al. Exome sequencing in the clinical diagnosis of sporadic or familial cerebellar ataxia. JAMA Neurol. 2014;71(10):1237-46.
- Fogel BL. Autosomal-recessive cerebellar ataxias. Handb Clin Neurol. 2018;147:187-209.
- Hadjivassiliou M, Grünewald RA, Sanders DS, et al. Effect of gluten-free diet on cerebellar MR spectroscopy in gluten ataxia. Neurology. 2017;89(7):705-9.
- Mitoma H, Hadjivassiliou M, Honnorat J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias. 2015;2:14.
- Lou JS, Goldfarb L, McShane L, et al. Use of buspirone for treatment of cerebellar ataxia. An open-label study. Arch Neurol. 1995;52(10):982-8.
- Trouillas P, Xie J, Adeleine P, et al. Buspirone, a 5-hydroxytryptamine1A agonist, is active in cerebellar ataxia. Results of a double-blind drug placebo study in patients with cerebellar cortical atrophy. Arch Neurol. 1997;54(6):749-52.
- Zesiewicz TA, Wilmot G, Kuo SH, et al. Comprehensive systematic review summary: treatment of cerebellar motor dysfunction and ataxia. Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018;90(10):464-71.
- Scoles DR, Pulst SM. Antisense therapies for movement disorders. Mov Disord. 2019;34(8):1112-9.
Engelsk sammanfattning
Ataxias constitute a group of heterogeneous diseases with overlapping symptoms. The clinical investigation should primarily seek for treatable conditions such as neurometabolic disorders and autoimmune diseases. Rapid progression is often characteristic for paraneoplastic cerebellar degeneration, autoimmune diseases or multiple system atrophy (MSA). The rapid development of massive parallel DNA sequencing and its increased accessibility have enabled for improved diagnostic resolution of patients. A diagnosis based on the etiology is crucial for prognosis and treatment of ataxias. In hereditary forms, the identification of causative genetic factors is essential for family planning.



















