






Lynchs syndrom innebär kraftigt ökad livstidsrisk för kolorektal cancer och endometriecancer vid lägre ålder än i normalpopulationen.
Ökad medvetenhet bland vårdpersonal om ärftliga cancertillstånd skulle öka chansen till att fler diagnostiserades innan de hunnit utveckla cancer.
Regelbunden övervakning av anlagsbärare med koloskopi och för kvinnor även transvaginalt ultraljud och endometriebiopsier rekommenderas i syfte att diagnostisera neoplastiska förändringar tidigt med målsättning att minska såväl morbiditet som mortalitet hos individer med Lynchs syndrom.
Ytterligare åtgärder i cancerpreventivt syfte, som kemoprevention och vaccination, är under utveckling.
Lynchs syndrom är ett av flera ärftliga syndrom där livstidsrisken för kolorektal cancer är kraftigt förhöjd. Syndromet orsakas av mutationer i eller i nära anslutning till DNA-reparationsgenerna (mismatch repair, MMR) MLH1, MSH2, MSH6 och PMS2, vilka kodar för proteiner som ska reparera defekt basparning under DNA-replikationen (Fakta 1) [1]. Nedärvningen är autosomalt dominant, vilket innebär att risken för ett barn till en individ med Lynchs syndrom att ärva anlaget är 50 procent. Innan mutationsbestämning var möjlig baserades diagnosen ofta på enbart kliniska kriterier och benämndes ärftlig icke-polypös kolorektal cancer (HNPCC; hereditary nonpolyposis colorectal cancer).
I Sverige får ca 6 000 personer årligen diagnosen kolorektal cancer, vilken utgör den andra vanligaste cancerrelaterade dödsorsaken [2]. Uppskattningsvis orsakas 3 procent av all kolorektal cancer av medfödda mutationer, däribland Lynchs syndrom [1].
Tumörspektrum och cancerrisk
Livstidsrisken för att insjukna i kolorektal cancer uppskattas för män med Lynchs syndrom vara mellan 54–74 procent och för kvinnor 30–52 procent [3]. Risken varierar beroende på vilken gen som är muterad [1]. Kolorektal cancer vid Lynchs syndrom diagnostiseras vid en lägre genomsnittlig ålder än kolorektal cancer i normalpopulationen [3]. Individer med Lynchs syndrom har i större utsträckning synkrona (flera samtidiga) tumörer och större risk för metakrona (nya) tumörer i samma organ än vad som är fallet vid sporadisk kolorektal cancer.
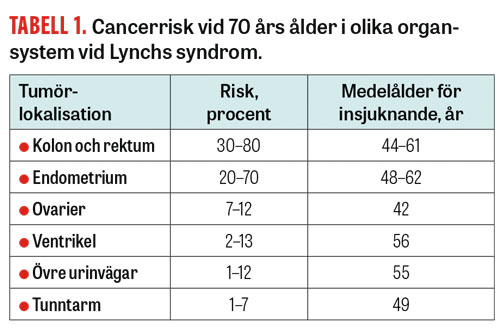
Lynchs syndrom är även kopplat till ökad risk för många andra typer av neoplasier förutom kolorektal cancer. Vanligast är endometriecancer; kvinnor med Lynchs syndrom har en livstidsrisk för endometriecancer på 20–70 procent [4]. Syndromet är även kopplat till ovarialcancer, urinvägscancer, ventrikelcancer och tunntarmscancer (Tabell 1) [4]. Det är därför viktigt att ha i åtanke att patienter med Lynchs syndrom kan debutera med annan tumörform än kolorektal cancer.
Utredningsgång vid misstänkt Lynchs syndrom
Historisk utredningsgång. Enbart kliniska kriterier tillämpades innan molekylärgenetisk diagnostik fanns att tillgå [1]. Diagnostik baserad enbart på dessa kriterier leder till underdiagnostik, eftersom många kan sakna typisk familjehistorik, vilket är fallet vid mutationer med nedsatt sjukdomspenetrans [1] eller där släktingar avlidit i ung ålder av andra omständigheter. Alla individer har inte heller tillgång till en utförlig medicinsk familjehistorik av olika sociala skäl.
Utredningsgång i nuläget. Individer som misstänks ha Lynchs syndrom själva eller i familjen bör remitteras till en klinisk genetisk mottagning för genetisk vägledning och ställningstagande till vidare genetisk diagnostik. En detaljerad familjeanamnes utgör det första diagnostiska verktyget. Svensk förening för medicinsk genetik har tagit fram riktlinjer (Fakta 2) för när ärftlighetsutredning av kolorektal cancer är indicerad [4].
Utöver kliniska kriterier finns även möjlighet att undersöka om det föreligger immunhistokemisk förlust av MMR-protein och DNA-markörer som mikrosatellitinstabilitet (MSI) i tumörvävnaden; dessa fenomen förekommer i stor utsträckning i tumörvävnad vid Lynchs syndrom men kan ibland ses i sporadiska tumörer [5].
Sekvensering av MMR-generna från lymfocyt-DNA (blodprov) är i dag praxis som diagnostisk metod för att kartlägga de Lynch-specifika gener som vi i nuläget känner till [4]. Fördelen med dessa metoder är att de anses ha hög specificitet. Nackdelen är dock att de kan ge svårtolkade resultat, eftersom man inte alltid kan bedöma den kliniska relevansen av en mutation, dvs om den är sjukdomsalstrande eller inte.
Framtida utredningsgång. Uppskattningsvis förbises upp till 25 procent av alla fall av cancer associerad till Lynchs syndrom, främst kolorektal cancer och endometriecancer [5, 6]. Eftersom det i dagsläget föreligger såväl fördröjd diagnostik som underdiagnostik av Lynchs syndrom, pågår diskussioner om att undersöka all kolorektal cancer och endometriecancer med avseende på Lynchs syndrom med immunhistokemisk screening av tumörvävnad följd av molekylärgenetisk diagnostik [5]. Fördelar med ett sådant förfarande skulle vara att fler individer med Lynchs syndrom skulle upptäckas, att diagnosen skulle ställas tidigare och att presymtomatisk genetisk diagnostik av släktingar skulle kunna erbjudas.
I Storbritannien publicerade National Institute for Health and Care Excellence (NICE) nyligen rekommendationer om att immunhistokemi avseende Lynchs syndrom ska utföras vid all nydiagnostiserad kolorektal cancer, och man konstaterade att detta är kostnadseffektivt [7]. American Society of Colon and Rectal Surgeons [8] rekommenderar i stället att all kolorektal cancer hos individer med debut före 70 års ålder ska undersökas med immunhistokemi, och om patienten i fråga är äldre får familjehistoriken vara avgörande. Det slutliga syftet är att detta inte bara ska identifiera individer med Lynchs syndrom utan också i förlängningen förebygga kolorektal cancer hos anlagsbärande släktingar.
I Sverige publicerade SBU (Statens beredning för medicinsk och social utvärdering) en rapport 2016 som påvisar att testning av personer yngre än 50 år med nydiagnostiserad kolorektal cancer och deras nära anhöriga innebär en låg till måttlig kostnad per hälsoeffekt (QALY) jämfört med att inte testa [9].
Det finns således mycket som talar för att införa utökad diagnostik för att hitta individer med Lynchs syndrom i Sverige. Nackdelar med ett sådant förfarande kan vara att det i dag saknas kunskap och resurser i vården i form av genetisk vägledning och uppföljningsrutiner för att ta hand om konsekvenserna av ett positivt test [5]. Etiska perspektiv som inkluderar patientens samtycke till sådan diagnostik och psykosocial påverkan av ett positivt test bör också beaktas, eftersom det inte bara berör den enskilda individen utan hela släkten. Det kan också påverka familjebildning och innebära en psykologisk belastning att leva med vetskapen om en signifikant ökad risk för cancer [5].
Genetisk vägledning. Syftet med den genetiska vägledningen är att bedöma risk att insjukna i cancer för individen och andra familjemedlemmar och att informera om förutsättningarna kring anlagsbärartest och möjligheterna till cancerförebyggande åtgärder. Genetisk vägledning innefattar också i förekommande fall diskussion kring eventuell framtida fosterdiagnostik/preimplantorisk genetisk diagnostik.
Utredningen innefattar noggrann familjeanamnes och kartläggning av vilka i släkten som insjuknat i cancer och ålder för diagnos. För närvarande saknas säkra metoder för att bedöma absolut cancerrisk.I nuläget kalkyleras risken främst utifrån familjehistorik och typ av mutation, där även förvärvade riskfaktorer som övervikt och rökning måste tas med i kalkylen. Därför bör denna patientgrupp aktivt erbjudas stöd vad gäller viktreduktion och rökavvänjning [10].
Uppföljning efter diagnos
Efter onkogenetisk utredning remitteras individen vidare till en specialistmottagning för uppföljning av riskorgan, i första hand kolon, rektum och uterus (Tabell 2) [4]. Vid mottagningen informeras individen om möjliga kontrollprogram. Övriga riskfaktorer för cancer kartläggs, t ex rökning och övervikt.
Endoskopisk övervakning. Nationella riktlinjer rekommenderar koloskopi varje till vartannat år med start vid 20–25 års ålder [4]. Dessa rekommendationer kan sannolikt komma att modifieras framöver och bli mer individanpassade. Ett exempel är att koloskopiintervallen skulle kunna komma att variera mer än i dag beroende på den enskildes uppskattade risk för kolorektal cancer baserat på bl a vilken gen som är muterad, hur familjehistorien avseende cancer ser ut, rökvanor och tidigare cancer i den egna sjukhistorien. Även ålder vid start av skopikontroller kan på samma grunder sannolikt individualiseras mer än vad som sker i dag.
En studie [11] från 2006 utvärderade övervakningsprogrammet i Nederländerna (som introducerades på sent 1980-tal) genom att analysera mortalitet i kolorektal cancer före och efter 1990. Man fann att mortaliteten för dessa patienter sjönk över tid (70 procent i standardiserad mortalitetskvot; P < 0,001), vilket antyder att effektiviteten av ett sådant övervakningsprogram är hög.
Risken för kolorektal cancer vid Lynchs syndrom anses inte vara tillräckligt hög för att rekommendera primärprofylaktisk kolektomi i förebyggande syfte. Vid koloncancer rekommenderas att hela kolon avlägsnas oavsett lokalisation, eftersom risken för metakron cancer är hög. Vid cancer i rektum görs även proktektomi, men vid koloncancer behåller man rektum och anlägger en ileorektal anastomos. Segmentresektion kan övervägas hos äldre individer och andra med hög operationsrisk. Oavsett kirurgiskt ingrepp är fortsatt postoperativ endoskopisk övervakning nödvändig så länge någon del av kolon eller rektum finns kvar [1].
Eftersom polyper kan vara ett förstadium till kolorektal cancer vid Lynchs syndrom är det viktigt att identifiera dessa. Polyper som sannolikt är förstadier till cancer kan vara av två slag: dels adenom, dels s k sågtandande (hyperplastiska) polyper. Sågtandade polyper är vanligen belägna i högerkolon. De är flacka och därmed svåra att upptäcka. Därför är det speciellt viktigt med noggrann undersökning av högerkolon med bra endoskopisk kvalitet där väl rengjord tarm är en mycket viktig förutsättning för att detta ska uppnås.
Cancerutvecklingen anses ske via liknande mekanismer som leder till mikrosatellitinstabilitet [13]. Eftersom de sågtandade polyperna ofta är svårare att upptäcka än adenom diskuteras om den endoskopiska uppföljningen av dessa individer bör ske med hjälp av mer avancerade tekniker än den som är rutin i dag [14]. Detta tillsammans med att erfarenhet av att diagnostisera flacka polyper är av stor vikt talar för att den endoskopiska övervakningen av Lynchs syndrom, i den mån det är möjligt, bör centraliseras.
Övrig övervakning. Utöver kontrollprogram för kolorektal cancer saknas entydig evidens vad gäller uppföljning av övriga riskorgan. På grund av osäkerheten kring möjligheten att upptäcka cancer i tid med gynekologisk övervakning erbjuds därför kvinnor som avslutat familjebildningen profylaktisk hysterektomi med samtidig salpingooforektomi [6]. Risken för ovarialcancer hos patienter med Lynchs syndrom är 7–12 procent [4] och är således en mer sällsynt manifestation. Ovarialcancer har emellertid sämre prognos och är mer svårdiagnostiserad än kolorektal cancer, vilket motiverar profylaktisk kirurgi [15]. Emellertid pågår diskussionen om huruvida denna strategi bör överges till förmån för ett förbättrat kontrollprogram [15].
Kvinnor med Lynchs syndrom som inte är hysterektomerade erbjuds rutinmässigt gynekologisk övervakning med avseende på endometrie- och ovarialcancer, med start vid 30–35 års ålder och med kontroll- intervall varje till vartannat år, även om startåldern till stor del beror på åldern hos den yngsta insjuknade släktingen. Övervakningen innefattar i dagsläget transvaginalt ultraljud, gynekologisk undersökning och endometriebiopsier [4].
Tanken bakom övervakning är att diagnostisera förstadier till endometrie- och ovarialcancer. Evidensen för övervakning avseende gynekologisk cancer är dock inte lika stark som den för kolorektal cancer. De studier som gjorts på området har inte kunnat påvisa någon större effektivitet vad gäller detektion av premaligna lesioner [6]. De fåtal studier som har kunnat påvisa någon effektivitet menar att utfallet snarare beror på att endometriecancer associerad till Lynchs syndrom i sig har bättre prognos än sporadisk endometriecancer. Frånsett tidigare insjuknandeålder skiljer sig inte övriga tumörkarakteristika, som tumörhistologi, vid endometriecancer associerad till Lynchs syndrom nämnvärt från den sporadiska motsvarigheten [16].
Kvinnor som går på regelbundna kontroller har ofta bättre kontakt med vården och är mer benägna att söka när de får symtom som kan härledas till gynekologisk malignitet [17]. Det råder delade meningar om vilka metoder som lämpar sig bäst för övervakning, likaså om vid vilken ålder kontroller bör starta och med vilket kontrollintervall. Emellertid är det svårt att göra studier under en längre tid, eftersom många kvinnor genomgår profylaktisk operation. Det är heller inte möjligt att göra prospektiva studier av etiska skäl. Det finns också en psykologisk aspekt att ta hänsyn till vad gäller obehag vid undersökningarna [6].
Övervakning erbjuds dock i nuläget till alla som inte är hysterektomerade, eftersom det anses vara oetiskt att inte erbjuda denna riskgrupp kontroller. I nuläget motiveras profylaktisk hysterektomi av att det inte finns ett etablerat säkert gynekologiskt övervakningsprogram [15].
I dag baseras uppföljning av annan malignitet än kolorektal cancer och endometriecancer på förekomst i familjehistoriken [4]. Även genotypen påverkar risken; t ex har MSH2-bärare högre risk för extrakoloniska maligniteter än bärare av övriga genotyper [6]. Kvinnliga MSH6-bärare löper högre risk för endometriecancer än bärare av andra genotyper [6]. Det är viktigt för patient och vårdpersonal att vara uppmärksam på varningssymtom som kan härledas till associerade cancerformer.
Om utökad uppföljning anses motiverad, sker denna med avseende på ventrikel- och tunntarmscancer i form av gastroskopi och/eller magnetkameraundersökning av tunntarm, och uppföljning med avseende på cancer i urinvägarna i form av cystoskopi och urincytologi.
Framtida cancerprofylax vid Lynchs syndrom
Utöver profylaktisk kirurgi samt endoskopisk och gynekologisk övervakning finns flertalet andra cancerpreventiva åtgärder under utveckling.
Eftersom man i observationsstudier noterade en skyddande effekt vid regelbunden användning av acetylsalicylsyra (ASA) med avseende på kolorektal cancer genomfördes en randomiserad placebokontrollerad studie (CAPP2) i syfte att utvärdera effekten vid Lynchs syndrom. Studien påvisade en incidensminskning av kolorektal cancer med 63 procent hos studiedeltagare som intagit 600 mg i upp till 4 år [18]. Intressant nog sågs även en signifikant minskning av andra maligniteter associerade till Lynchs syndrom.
Dosen 600 mg ASA per dag som långtidsprofylax är dock associerad med biverkningar, varav vissa kan vara allvarliga. Ett alternativ kan visa sig vara 5-aminosalicylsyra (5-ASA), ett preparat utan ASA:s biverkningsprofil med ökad blödningsrisk, som i dag används vid behandling av inflammatorisk tarmsjukdom och som också har visat sig minska risken för kolorektal cancer [19]. Dock saknas ännu randomiserade studier vid Lynchs syndrom.
Eftersom det ännu inte finns tillräckligt med data vad gäller optimal dosering och nödvändig behandlingslängd för kemopreventiv behandling, bör man invänta resultatet av pågående studier innan man börjar använda något av dessa läkemedel inom rutinsjukvården.
Maligniteter associerade till Lynchs syndrom har en tendens till bättre prognos vad gäller mortalitet än de sporadiska motsvarigheterna [20]. En bidragande orsak till detta kan vara att immunförsvaret aktiveras och ger upphov till uttalad lymfocytinfiltration i tumörvävnaden. Anledningen till detta fenomen tros vara att defekt MMR orsakar ett skifte i DNA-läsramen, vilket i sin tur ger upphov till immunogena peptider som stimulerar lymfocytinfiltration.
Denna mekanism har i sin tur föreslagits som en potentiell angreppspunkt för ett vaccin att användas i syfte att både förebygga och behandla redan etablerad cancer. Kolorektal cancer associerad till Lynchs syndrom svarar däremot inte lika bra på cytostatika som sporadisk kolorektal cancer [20].
Deskriptiv studie av vanligaste orsakerna till diagnos
Mottagningen för ärftlig mag–tarmcancer, patientflöde ärftlig cancer vid Karolinska universitetssjukhuset Solna följer upp majoriteten av de individer i Stockholms läns landsting som har Lynchs syndrom. I syfte att kvalitetsgranska verksamheten genomförde artikelförfattarna under 2015 ett projekt inom ramen för ett examensarbete på läkarprogrammet, nämligen en genomgång av alla patienter som någon gång haft ett besök vid denna mottagning (Tabell 3).
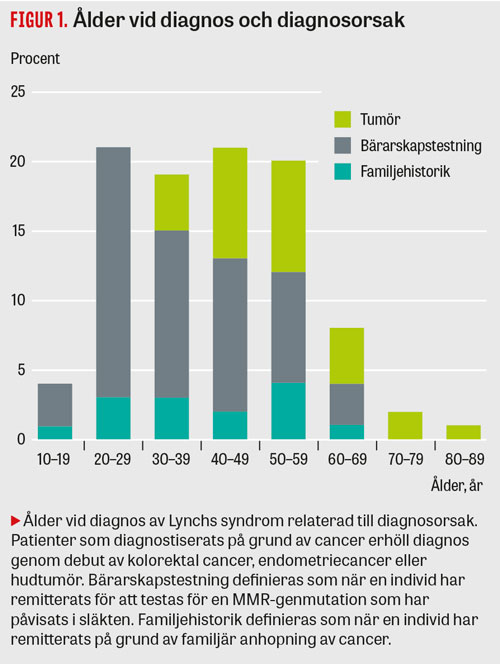
En av huvudfrågeställningarna i den deskriptiva studien var att kartlägga patienternas väg till diagnosen Lynchs syndrom. Inklusionskriterier var minst ett besök på mottagningen och en molekylärgenetiskt bekräftad diagnos. Data från 1980 till och med 2015 på 121 patienter inhämtades via genomgång av journaler. Nästan var tredje patient (49/121) utreddes och diagnostiserades med Lynchs syndrom i samband med en cancerdiagnos.
Dessutom observerades att äldre individer (36–80 år) i större utsträckning än yngre diagnostiserades med Lynchs syndrom i samband med en cancerdiagnos (Figur 1). Yngre individer får anlagsbärartest oftare än äldre på grund av antingen familjehistorien eller att MMR-mutationen redan var känd i familjen. Inga könsskillnader förelåg vad gäller vägen till diagnos.
Arbetet gjordes inom ramen för kvalitetsgranskning av verksamheten och berör sjukvårdshuvudmannens ansvar för en specifik patientgrupp. Formellt krävs inte etiktillstånd för kvalitetsgranskning (etikprövningsnämndens utlåtande, dnr 2015/307-31/1).
Sammanfattningsvis visar denna genomgång att en stor andel individer får sitt anlagsbärarskap för Lynchs syndrom fastställt först när de drabbats av cancer. Dessa individer har haft otillräcklig kunskap före insjuknandet om sin genetiskt förhöjda risk, vilket indikerar att det föreligger ett stort mörkertal när det gäller individer/familjer med Lynchs syndrom och tyder på att det behövs bättre strategi och kunskap på nationell nivå för att diagnostisera patienter med Lynchs syndrom.
Framtida målsättningar
En framtida målsättning måste vara att andelen individer med presymtomatisk diagnos ökar. En annan viktig målsättning är att optimera sjukvårdens rutiner för att identifiera potentiella fall av Lynchs syndrom, i första hand bland alla de patienter som drabbas av kolorektal cancer och endometriecancer i Sverige, men också genom att bli bättre på att penetrera och se mönster i familjehistorien.
För att uppnå dessa mål måste kunskapen om ärftliga cancersyndrom öka genom utbildning och information såväl i vården (primärvård och specialistvård) som bland allmänheten. Dessutom måste berörda verksamheter organiseras och resursförstärkas så att det är möjligt att testa alla fall av kolorektal cancer och endometriecancer avseende Lynchs syndrom.
Vidare måste man förbereda för att kunna erbjuda genetisk vägledning och kontroller till en betydligt större population patienter med Lynchs syndrom än den vi i dag känner till. Även om man sannolikt aldrig kommer att uppnå 100-procentig detektionsgrad, eftersom detta skulle fordra screening av hela befolkningen, kan man med relativt enkla åtgärder som en detaljerad anamnes avseende i första hand kolorektala polyper, kolorektal cancer och endometriecancer i såväl individ- som familjehistorien öka chansen att upptäcka dessa individer.
Eftersom utvecklingen går snabbt, med förbättrade och individanpassade kontrollprogram och inom en snar framtid sannolikt möjlighet till kemoprevention, är det viktigt att vi blir bättre på att identifiera anlagsbärare, så att fler kan erbjudas olika typer av åtgärder i cancerpreventivt syfte.
Potentiella bindningar eller jävsförhållanden: Inga uppgivna.
Fakta 1. Genetik vid Lynchs syndrom
Lynchs syndrom ärvs autosomalt dominant och orsakas av mutationer i eller i nära anslutning till DNA-reparationsgenerna (mismatch repair, MMR) MLH1, MSH2, MSH6 och PMS2. Dessa gener kodar för proteiner som reparerar defekt basparning under DNA-replikationen.
Mutationer i dessa gener orsakar skador i framför allt icke-kodande delar av DNA, vilket ger upphov till s k mikrosatellitinstabilitet (MSI) och skador i tumörsuppressorgener med korta repetitiva DNA-sekvenser i kodande regioner.
Tumörer som orsakas av Lynchs syndrom kännetecknas därför av mikrosatellitinstabilitet, vilket tidigare har ansetts vara så specifikt att det har använts diagnostiskt.
Fakta 2. Indikationer för ärftlighetsutredning
Remiss bör skickas till mottagning för klinisk genetik i följande fall:
- Individ/familj där någon har insjuknat i kolorektal cancer och/eller endometriecancer före 50 års ålder.
- Individ/familj med två eller flera personer med kolorektal cancer.
- Individ/familj med två eller flera personer med maligna tumörer associerade till ärftlig kolorektal cancer.
- Individ med metakron/synkron malign tumör associerad till ärftlig kolorektal cancer.
- Individ med 10 eller fler kolorektala polyper.
Referenser
- Jass JR. Hereditary non-polyposis colorectal cancer: the rise and fall of a confusing term. World J Gastroenterol. 2006;12(31):4943-50.
- Sveriges officiella statistik. Cancer incidence in Sweden 2007. Stockholm: Socialstyrelsen; 2008. Artikelnr 2008-125-16.
- Bellcross CA, Bedrosian SR, Daniels E, et al. Implementing screening for Lynch syndrome among patients with newly diagnosed colorectal cancer: summary of a public health/clinical collaborative meeting. Genet Med. 2012;14(1):152-62.
- Ärftlig kolorektalcancer – utredning, uppföljning och omhändertagande. Stockholm: Svensk förening för medicinsk genetik, Arbetsgruppen för cancergenetiska mottagningar; 2012.
- de la Chapelle A, Hampel H. Clinical relevance of microsatellite instability in colorectal cancer. J Clin Oncol. 2010;28(20):3380-7.
- Vasen HF, Blanco I, Aktan-Collan K, et al; Mallorca group. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut. 2013;62(6):812-23.
- Molecular testing strategies for Lynch syndrome in people with colorectal cancer. NICE Diagnostics guidance (DG27). London: National Institute of Health and Care Excellence; 2017. https://www.nice.org.uk/guidance/dg27
- Herzig DO, Buie WD, Weiser MR, et al. Clinical practice guidelines for the surgical treatment of patients with Lynch syndrome. D. Dis Colon Rectum 2017;60(2):137-43.
- SBU kommenterar. Utvärdering av diagnostiska strategier vid ärftlig tjocktarmscancer (Lynch syndrom). Stockholm: Statens beredning för medicinsk och social utvärdering; 2016.
- Pande M, Lynch PM, Hopper JL, et al. Smoking and colorectal cancer in Lynch syndrome: results from the Colon Cancer Family Registry and the University of Texas M.D. Anderson Cancer Center. Clin Cancer Res. 2010;16(4):1331-9.
- de Jong AE, Hendriks YM, Kleibeuker JH. Decrease in mortality in Lynch syndrome families because of surveillance. Gastroenterology. 2006;130(3):665-71.
- Stoffel EM, Turgeon DK, Stockwell, et al; Great Lakes-New England Clinical Epidemiology and Validation Center of the Early Detection Research Network. Missed adenomas during colonoscopic surveillance in individuals with Lynch Syndrome (hereditary nonpolyposis colorectal cancer). Cancer Prev Res (Phila). 2008;1(6):470-5.
- Jass JR, Iino H, Ruszkiewicz A, et al. Neoplastic progression occurs through mutator pathways in hyperplastic polyposis of the colorectum. Gut. 2000;47(1):43-9.
- Oxentenko AS, Smyrk TC. Interval colon cancer in a Lynch syndrome patient under annual colono-scopic surveillance: a case for advanced imaging techniques? BMC Gastroenterol. 2012;12:50.
- Dębniak T, Gromowski T, Scott RJ, et al. Management of ovarian and endometrial cancers in women belonging to HNPCC carrier families: review of the literature and results of cancer risk assessment in Polish HNPCC families. Hered Clin Cancer Pract. 2015;13(1):3.
- Meyer LA, Broaddus RR, Lu KH. Endometrial cancer and Lynch syndrome: clinical and pathologic considerations. Cancer Control. 2009;16(1):14-22.
- Stuckless S, Green J, Dawson L, et al. Impact of gynecological screening in Lynch syndrome carriers with an MSH2 mutation. Clin Genet. 2013;83(4):359-64.
- Burn J, Mathers JC, Bishop DT. Chemoprevention in Lynch syndrome. Fam Cancer. 2013;12(4):707-18.
- Velayos FS, Terdiman JP, Walsh JM. Effect of 5-aminosalicylate use on colorectal cancer and dysplasia risk: a systematic review and metaanalysis of observational studies. Am J Gastroenterol. 2005;100(6):1345-53.
- von Knebel Doeberitz M, Kloor M. Towards a vaccine to prevent cancer in Lynch syndrome patients. Fam Cancer. 2013;12(2):307-12.
Summary
Lynch syndrome is a major cause of monogenetic familial colorectal cancer
Individuals with a history of colorectal or endometrial cancer younger than 50 years of age or with relatives younger than 50 years of age with a history of any of these malignances should be referred to clinical genetics for further diagnostic examinations and genetic counseling. Lynch syndrome is caused by mutations in mismatch repair genes and implicates an increased risk for colorectal cancer as well as endometrial cancer. Routine surveillance for this group of individuals regarding colorectal cancer by means of colonoscopy and endometrial cancer by means of transvaginal ultrasound as well as endometrial biopsies is recommended annually or biennially. Several preventive measures are under development, such as chemoprevention and vaccination. During 2015 we investigated reasons for diagnosis among those registered at Karolinska University Hospital. We found that a substantial part of this group of individuals was diagnosed in conjunction with their diagnosis of cancer; a prerequisite in order to offer at-risk individuals preventive measures is to improve identification of these individuals and offer them presymptomatic genetic testing in order to identify predisposing mutations.


















